To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Intermolecular forceIn physics, chemistry, and biology, intermolecular forces are forces that act between stable molecules or between functional groups of macromolecules. Intermolecular forces (aka van der Waal's forces) include momentary attractions between molecules, diatomic free elements, and individual atoms. They differ from covalent and ionic bonding in that they are not stable, but are caused by momentary polarization of particles. Because electrons have no fixed position in the structure of an atom or molecule, but rather are distributed in a probabilistic fashion based on quantum probability, there is a positive chance that the electrons are not evenly distributed and thus their electrical charges are not evenly distributed. See Schrodinger Equation for the theories on wave functions and descriptions of position and velocity of quantum particles. In general one distinguishes short and long range van der Waal's forces. The former are due to intermolecular exchange and charge penetration. They fall off exponentially as a function of intermolecular distance R and are repulsive for interacting closed-shell systems. In chemistry they are well known, because they give rise to steric hindrance, also known as Born or Pauli repulsion. Long range forces fall off with inverse powers of the distance, R-n, typically 3 ≤ n ≤ 10, and are mostly attractive. The sum of long and short range forces gives rise to a minimum, referred to as Van der Waal minimum. The position and depth of the Van der Waal's minimum depends on distance and mutual orientation of the molecules. Additional recommended knowledge
General theoryThis is because before the advent of quantum mechanics the origin of intermolecular forces was not well understood. Especially the causes of hard sphere repulsion, postulated by Van der Waals, and the possibility of the liquefaction of noble gases were difficult to understand. Soon after the formulation of quantum mechanics, however, all open questions regarding intermolecular forces were answered, first by S.C. Wang and then more completely and thoroughly by Fritz London. The quantum mechanical basis for the majority of intermolecular effects is contained in a nonrelativistic energy operator, the molecular Hamiltonian. This operator consists only of kinetic energies and Coulomb interactions. Usually one applies the Born-Oppenheimer approximation and considers the electronic (clamped nuclei) Hamilton operator only. For very long intermolecular distances the retardation of the Coulomb force (first considered in 1948 for intermolecular forces by Hendrik Casimir and Dirk Polder) may have to be included. Sometimes, e.g., for interacting paramagnetic or electronically excited molecules, electronic spin and other magnetic effects may play a role. In this article, however, retardation and magnetic effects will not be considered. We will distinguish four fundamental interactions:
Perturbation theoryThe last three of the fundamental interactions are most naturally accounted for by Rayleigh-Schrödinger perturbation theory (RS-PT). In this theory—applied to two monomers A and B—one uses as unperturbed Hamiltonian the sum of two monomer Hamiltonians, while the perturbation is where qi indicates the charge (in units e of elementary charge) of a particle of monomer A; qj belongs to monomer B. For electrons we take q = -1, for a nucleus we take q equal to its atomic number Z. The quantity rij is the distance between particle i and particle j. In this equation and further in this article atomic units are used. Perturbation theory is based on expansions of perturbed states in terms of unperturbed states (eigenstates of H(0)). In the present case the unperturbed states are products Supermolecular approachThe early theoretical work on intermolecular forces was invariably based on RS-PT and its antisymmetrized variants. However, since the beginning of the 1990s it has become possible to apply standard quantum chemical methods to pairs of molecules. This approach is referred to as the supermolecule method. In order to obtain reliable results one must include electronic correlation in the supermolecule method (without it dispersion is not accounted for at all), and take care of the basis set superposition error. This is the effect that the atomic orbital basis of one molecule improves the basis of the other. Since this improvement is distance dependent, it gives easily rise to artefacts. Supermolecule calculations must be performed with very high precision, because the problem, known as weighing the captain, arises here. First we weigh the ship with the captain aboard (total energy of molecules in interaction) and then we weigh the ship with the captain ashore (total energy of molecules at an infinite distance apart); the difference gives the captain's weight. This parable is due to the late Charles Coulson. To understand it we must remember that the total energy of molecules is six to seven orders of magnitude larger than a typical intermolecular interaction. That is, the significant digits in the results of supermolecule calculations start to appear beyond the sixth or seventh decimal place. A disadvantage of the supermolecule method is that it yields the interaction as a lump sum. It does not give an interaction energy separated in the four fundamental contributions mentioned above. Therefore, we shall not discuss the supermolecule method any further in this article. ExchangeThe monomer functions ΦnA and ΦmB are antisymmetric under permutation of electron coordinates (i.e., they satisfy the Pauli principle), but the product states are not antisymmetric under intermolecular exchange of the electrons. An obvious way to proceed would be to introduce the intermolecular antisymmetrizer In the second place the projected excited states become linearly dependent and the choice of a linearly independent subset is not apparent. In the late 1960s the Eisenschitz-London approach was revived and different rigorous variants of symmetry adapted perturbation theory were developed. (The word symmetry refers here to permutational symmetry of electrons). The different approaches shared a major drawback: they were very difficult to apply in practice. Hence a somewhat less rigorous approach (weak symmetry forcing) was introduced: apply ordinary RS-PT and introduce the intermolecular antisymmetrizer at appropriate places in the RS-PT equations. This approach leads to feasible equations, and, when electronically correlated monomer functions are used, weak symmetry forcing is known to give reliable results.[2][3] The first-order (most important) energy including exchange is in almost all symmmetry-adapted perturbation theories given by the following expression The main difference between covalent and non-covalent forces is the sign of this expression. In the case of chemical bonding this interaction is attractive (for certain electron-spin state, usually spin-singlet) and responsible for large bonding energies—on the order of a hundred kcal/mol. In the case of intermolecular forces between closed shell systems, the interaction is strongly repulsive and responsible for the "volume" of the molecule (see Van der Waals radius). Roughly speaking, the exchange interaction is proportional to the differential overlap between Φ0A and Φ0B. Since the wavefunctions decay exponentially as a function of distance, the exchange interaction does too. Hence the range of action is relatively short, which is why exchange interactions are referred to as short range interactions.
Electrostatic interactionsBy definition the electrostatic interaction is given by the first-order Rayleigh-Schrödinger perturbation (RS-PT) energy (without exchange): Let the clamped nucleus α on A have position vector Rα, then its charge times the Dirac delta function, Zα δ(r-Rα), is the charge density of this nucleus. The total charge density of monomer A is given by with the electronic charge density given by an integral over nA - 1 primed electron coordinates: An analogous definition holds for the charge density of monomer B. It can be shown that the first-order quantum mechanical expression can be written as which is nothing but the classical expression for the electrostatic interaction between two charge distributions. This shows that the first-order RS-PT energy is indeed equal to the electrostatic interaction between A and B. Multipole expansionAt present it is feasible to compute the electrostatic energy without any further approximations other than those applied in the computation of the monomer wavefunctions. In the past this was different and a further approximation was commonly introduced: VAB was expanded in a (truncated) series in inverse powers of the intermolecular distance R. This yields the multipole EXPANSION of the electrostatic energy. Since its concepts still pervade the theory of intermolecular forces, we will present it here. In this article the following expansion is proved with the Clebsch-Gordan series defined by and the irregular solid harmonic is defined by The function YL,M is a normalized spherical harmonic, while
Insertion of this expansion into the first-order (without exchange) expression gives a very similar expansion for the electrostatic energy, because the matrix element factorizes, with the permanent multipole moments defined by We see that the series is of infinite length, and, indeed, most molecules have an infinite number of non-vanishing multipoles. In the past, when computer calculations for the permanent moments were not yet feasible, it was common to truncate this series after the first non-vanishing term. Which term is non-vanishing, depends very much on the symmetry of the molecules constituting the dimer. For instance, molecules with an inversion center such as a homonuclear diatomic (e.g., molecular nitrogen N2), or an organic molecule like ethene (C2H4) do not posses a permanent dipole moment (l=1), but do carry a quadrupole moment (l=2). Molecules such a hydrogen chloride (HCl) and water (H2O) lack an inversion center and hence do have a permanent dipole. So, the first non-vanishing electrostatic term in, e.g., the N2—H2O dimer, is the lA=2, lB=1 term. From the formula above follows that this term contains the irregular solid harmonic of order L = lA + lB = 3, which has an R-4 dependence. But in this dimer the quadrupole-quadrupole interaction (R-5) is not unimportant either, because the water molecule carries a non-vanishing quadrupole as well. When computer calculations of permanent multipole moments of any order became possible, the matter of the convergence of the multipole series became urgent. It can be shown that, if the charge distributions of the two monomers overlap, the multipole expansion is formally divergent. Ionic interactionsIt is debatable whether ionic interactions are to be seen as intermolecular forces, some workers consider them rather as special kind of chemical bonding. The forces occur between charged atoms or molecules (ions). Ionic bonds are formed when the difference between the electron affinity of one monomer and the ionization potential of the other is so large that electron transfer from the one monomer to the other is energetically favorable. Since a transfer of an electron is never complete there is always a degree of covalent bonding. Once the ions (of opposite sign) are formed, the interaction between them can seen as a special case of multipolar attraction, with a 1/RAB distance dependence. Indeed, the ionic interaction is the electrostatic term with lA = 0 and lB = 0. Using that the irregular harmonics for L = 0 is simply and that the monopole moments and their Clebsch-Gordan coupling are (where qA and qB are the charges of the molecular ions) we recover—as to be expected—Coulomb's law For shorter distances, where the charge distributions of the monomers overlap, the ions will repel each other because of inter-monomer exchange of the electrons. Ionic compounds have high melting and boiling points due to the large amount of energy required to break the forces between the charged ions. When molten they are also good conductors of heat and electricity, due to the free or delocalised ions. Dipole-dipole interactionsDipole-dipole interactions, also called Keesom interactions or Keesom forces after Willem Hendrik Keesom, who produced the first mathematical description in 1921, are the forces that occur between two molecules with permanent dipoles. They result from the dipole-dipole interaction between two molecules. An example of this can be seen in hydrochloric acid:
The molecules are depicted here as two point dipoles. A point dipole is an idealization similar to a point charge (a finite charge in an infinitely small volume). A point dipole consists of two equal charges of opposite sign δ+ and δ-, which are a distance d apart. This distance d is so small that at any distance R from the point dipole it can be assumed that d/R >> (d/R)2. In this idealization the electrostatic field outside the charge distribution consists of one (R-3) term only, see this article. The electrostatic interaction between two point dipoles is given by the single term lA = 1 and lB = 1 in the expansion above. Obviously, no molecule is an ideal point dipole, and in the case of the HCl dimer, for instance, dipole-quadrupole, quadrupole-quadrupole, etc. interactions are by no means negligible (and neither are induction or dispersion interactions). Note that almost always the dipole-dipole interaction between two atoms is zero, because atoms rarely carry a permanent dipole, see atomic dipoles. Writing and similarly for B, we get the well-known expression As a numerical example we consider the HCl dimer depicted above. We assume that the left molecule is A and the right B, so that the z-axis is along the molecules and points to the right. Our (physical) convention of the dipole moment is such that it points from negative to positive charge. Note parenthetically that in organic chemistry the opposite convention is used. Since organic chemists hardly ever perform vector computations with dipoles, confusion hardly ever arises. In organic chemistry dipoles are mainly used as a measure of charge separation in a molecule. So, The value of μHCl is 0.43 (atomic units), so that at a distance of 10 bohr the dipole-dipole attraction is -3.698 10-4 hartree (-0.97 kJ/mol). If one of the molecules is neutral and freely rotating, the total electrostatic interaction energy becomes zero. (For the dipole-dipole interaction this is most easily proved by integrating over the spherical polar angles of the dipole vector, while using the volume element sinθ dθdφ). In gases and liquids molecules are not rotating completely freely—the rotation is weighted by the Boltzmann factor exp(-Edip-dip/kT), where k is the Boltzmann constant and T the absolute temperature. It was first shown by Lennard-Jones[4] that the temperature-averaged dipole-dipole interaction is Since the averaged energy has an R-6 dependence, it is evidently much weaker than the unaveraged one, but it is not completely zero. It is attractive, because the Boltzmann weighting favors somewhat the attractive regions of space. In HCl-HCl we find for T = 300 K and RAB = 10 bohr the averaged attraction -62 J/mol, which shows a weakening of the interaction by a factor of about 16 due to thermal rotational motion. Hydrogen bondingHydrogen bonding is an intermolecular interaction with a hydrogen atom being present in the intermolecular bond. This hydrogen is covalently (chemically) bound in one molecule, which acts as the proton donor. The other molecule acts as the proton acceptor. In the following important example of the water dimer, the water molecule on the right is the proton donor, while the one on the left is the proton acceptor:
The hydrogen atom participating in the hydrogen bond is often covalently bound in the donor to an electronegative atom. Examples of such atoms are nitrogen, oxygen, or fluorine. The electronegative atom is negatively charged (carries a charge δ-) and the hydrogen atom bound to it is positively charged. Consequently the proton donor is a polar molecule with a relatively large dipole moment. Often the positively charged hydrogen atom points towards an electron rich region in the acceptor molecule. The fact that an electron rich region exists in the acceptor molecule, implies already that the acceptor has a relatively large dipole moment as well. The result is a dimer that to a large extent is bound by the dipole-dipole force. For quite some time it was believed that hydrogen bonding required an explanation that was different from the other intermolecular interactions. However, reliable computer calculations that became possible during the 1980s have shown that only the four effects listed above play a role, with the dipole-dipole interaction being particularly important. Since the four effects account completely for the bonding in small dimers like the water dimer, for which highly accurate calculations are feasible, it is now generally believed that no other bonding effects are operative. Hydrogen bonds are found throughout nature. They give water its unique properties that are so important to life on earth. Hydrogen bonds between hydrogen atoms and nitrogen atoms of adjacent base pairs provide the intermolecular force that help more precisely bind together the two strands in a molecule of DNA. Hydrophobic effects between the double-stranded DNA and the surrounding aqueous environment, however, are more important in maintaining the DNA in its double stranded form. London dispersion forcesAlso called London forces, instantaneous dipole (or multipole) effects (spatially variable δ+) or Van der Waals forces, these involve the attraction between temporarily induced dipoles in nonpolar molecules (often disappear within an instant). This polarization can be induced either by a polar molecule or by the repulsion of negatively charged electron clouds in nonpolar molecules. An example of the former is chlorine dissolving in water: (+)(-)(+) (-) (+) [Permanent Dipole] H-O-H-----Cl-Cl [Induced Dipole] Note added by other author: Sketched is an interaction between the permanent dipole on water and an induced dipole on chlorine. The latter dipole is induced by the electric field offered by the permanent dipole of water (see field from an electric dipole). This permanent dipole-induced dipole interaction is referred to as induction (or polarization) interaction and is to be distinguished from the London dispersion interaction. The latter is sometimes described as an interaction between two instantaneous dipoles, see molecular dipole. The Cl2—Cl2 interaction that now follows is an example of a proper London dispersion interaction. (+) (-) (+) (-) [instantaneous dipole] Cl-Cl------Cl-Cl [instantaneous dipole] Note added by other author: It must be pointed out that the London interaction is not the only interaction between two chlorine molecules in the region where the overlap between the respective charge distributions may be neglected. Each chlorine molecule carries permanent multipole moments of even order, the first one being a permanent quadrupole moment (order 2). The interaction between two permanent multipole moments also contributes to the intermolecular force and the first term (quadrupole-quadrupole) is as important as the London dispersion force. London dispersion forces exist between all atoms. London forces are the only reason for rare-gas atoms to condense at low temperature. Quantum mechanical theory of dispersion forcesThe first explanation of the attraction between noble gas atoms was given by Fritz London in 1930.[5] He used a quantum mechanical theory based on second-order perturbation theory. The perturbation is the Coulomb interaction V between the electrons and nuclei of the two monomers (atoms or molecules) that constitute the dimer. The second-order perturbation expression of the interaction energy contains a sum over states. The states appearing in this sum are simple products of the excited electronic states of the monomers. Thus, no intermolecular antisymmetrization of the electronic states is included and the Pauli exclusion principle is only partially satisfied. London developed the perturbation V in a Taylor series in This Taylor expansion is known as the multipole expansion of V because the terms in this series can be regarded as energies of two interacting multipoles, one on each monomer. Substitution of the multipole-expanded form of V into the second-order energy yields an expression that resembles somewhat an expression describing the interaction between instantaneous multipoles (see the qualitative description above). Additionally an approximation, named after Albrecht Unsöld, must be introduced in order to obtain a description of London dispersion in terms of dipole polarizabilities and ionization potentials. In this manner the following approximation is obtained for the dispersion interaction Note that this final London equation does not contain instantaneous dipoles (see molecular dipoles). The "explanation" of the dispersion force as the interaction between two such dipoles was invented after London gave the proper quantum mechanical theory. See the authoritative work[6] for a criticism of the instantaneous dipole model and[7] for a modern and thorough exposition of the theory of intermolecular forces. The London theory has much similarity to the quantum mechanical theory of light dispersion, which is why London coined the phrase "dispersion effect" for the interaction that we described in this lemma. Anisotropy and non-additivity of intermolecular forcesConsider the interaction between two electric point charges at position
Consider three arbitrary point charges at distances r12, r13, and r23 apart. The total interaction U is additive; i.e., it is the sum
Again for molecules this can be different. Pretending that the interaction depends on distances only—but see above—the interaction of three molecules takes in general the form
where u(r12,r13,r23) is a non-additive three-body interaction. Such an interaction can be caused by exchange interactions, by induction, and by dispersion (the Axilrod-Teller triple dipole effect). See also
References
Categories: Intermolecular forces | Chemical bonding |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Intermolecular_force". A list of authors is available in Wikipedia. |



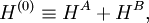
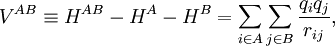
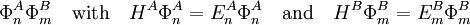
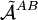 . But, as already noticed in 1930 by Eisenschitz and London,
. But, as already noticed in 1930 by Eisenschitz and London,![\big[ \tilde{\mathcal{A}}^{AB}, H^{(0)}\big] \ne 0 .](images/math/d/2/9/d29f9a88150d57a519f1ef3ccb864b89.png)
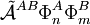
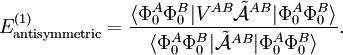
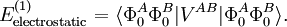
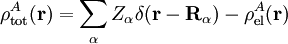
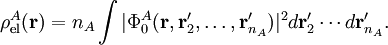
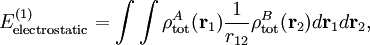
![V^{AB} = \sum_{\ell_A=0}^\infty \sum_{\ell_B=0}^\infty (-1)^{\ell_B} \binom{2\ell_A+2\ell_B}{2\ell_A}^{1/2} \sum_{M=-\ell_A-\ell_B}^{\ell_A+\ell_B} (-1)^{M} I_{\ell_A+\ell_B,-M}(\mathbf{R}_{AB})\; \left[\mathbf{Q}^{\ell_A} \otimes \mathbf{Q}^{\ell_B} \right]^{\ell_A+\ell_B}_M](images/math/8/5/f/85f1f7b9b51ad528bf20a30080fee0bf.png)
![\left[\mathbf{Q}^{\ell_A} \otimes \mathbf{Q}^{\ell_B} \right]^{\ell_A+\ell_B}_M \equiv \sum_{m_A=-\ell_A}^{\ell_A} \sum_{m_B=-\ell_B}^{\ell_B}\; Q_{m_A}^{\ell_A} Q_{m_B}^{\ell_B}\;\langle \ell_A, m_A; \ell_B, m_B| \ell_A+\ell_B, M \rangle.](images/math/3/c/0/3c0e59d08ab63939395a56800583b411.png)
![I_{L,M}(\mathbf{R}_{AB}) \equiv \left[\frac{4\pi}{2L+1}\right]^{1/2}\; \frac{Y_{L,M}(\widehat{\mathbf{R}}_{AB})}{R_{AB}^{L+1}}.](images/math/3/6/e/36e036079607f7b79e200c90cc512318.png)
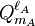 and
and 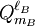 are spherical multipole moment operators. This expansion is manifestly in powers of 1/RAB.
are spherical multipole moment operators. This expansion is manifestly in powers of 1/RAB.
![\begin{align} E^{(1)}_\mathrm{electrostatic} = & \sum_{\ell_A=0}^\infty \sum_{\ell_B=0}^\infty (-1)^{\ell_B} \binom{2\ell_A+2\ell_B}{2\ell_A}^{1/2} \\ &\sum_{M=-\ell_A-\ell_B}^{\ell_A+\ell_B} (-1)^{M} I_{\ell_A+\ell_B,-M}(\mathbf{R}_{AB})\; \left[\mathbf{M}^{\ell_A} \otimes \mathbf{M}^{\ell_B} \right]^{\ell_A+\ell_B}_M, \end{align}](images/math/7/4/6/746ec3315e9317e677f9587002540afc.png)
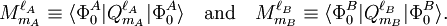
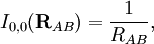
![M^{0_A}_0 = q_A,\quad M^{0_B}_0 = q_B \quad\hbox{and}\quad [\mathbf{M}^{0_A}\otimes \mathbf{M}^{0_A}]^0_0 = q_A q_B ,](images/math/2/4/7/2470d5dc366b298f5bdafc85f87e0703.png)
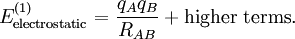
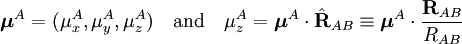
![E_{\mathrm{dip-dip}} = \frac{1}{R^{3}_{AB}}\left[ \boldsymbol{\mu}^A\cdot\boldsymbol{\mu}^B - 3 (\boldsymbol{\mu}^A\cdot \hat{\mathbf{R}}_{AB}) (\hat{\mathbf{R}}_{AB}\cdot \boldsymbol{\mu}^B) \right].](images/math/e/5/f/e5fdf9dce22f63f05ed21bac182aab48.png)
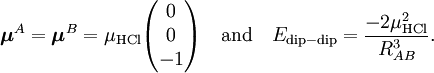
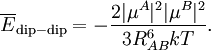
 , where
, where  between two atoms
between two atoms 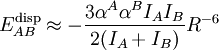
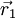 and
and 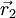 . By
. By
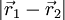 between
the particles. For molecules this is different. If we see a molecule as
a rigid 3-D body, it has 6 degrees of freedom (3 degrees for its
orientation and 3 degrees for its position in R3). The
interaction energy of two molecules (a dimer) in isotropic and
homogeneous space is in general a function of 2×6−6=6 degrees of freedom
(by the homogeneity of space the interaction does not depend on the
position of the center of mass of the dimer, and by the isotropy of
space the interaction does not depend on the orientation of the dimer).
The analytic description of the interaction of two arbitrarily shaped rigid
molecules requires therefore 6 parameters. (One often uses two
Euler angles per molecule, plus a dihedral angle, plus the distance.) The
fact that the intermolecular interaction depends on the orientation of the molecules
is expressed by stating that the potential is anisotropic. Since point charges are
by definition spherical symmetric, their interaction is isotropic. Especially in the
older literature, intermolecular interactions are regularly assumed to be
isotropic (e.g., the potential is described in
between
the particles. For molecules this is different. If we see a molecule as
a rigid 3-D body, it has 6 degrees of freedom (3 degrees for its
orientation and 3 degrees for its position in R3). The
interaction energy of two molecules (a dimer) in isotropic and
homogeneous space is in general a function of 2×6−6=6 degrees of freedom
(by the homogeneity of space the interaction does not depend on the
position of the center of mass of the dimer, and by the isotropy of
space the interaction does not depend on the orientation of the dimer).
The analytic description of the interaction of two arbitrarily shaped rigid
molecules requires therefore 6 parameters. (One often uses two
Euler angles per molecule, plus a dihedral angle, plus the distance.) The
fact that the intermolecular interaction depends on the orientation of the molecules
is expressed by stating that the potential is anisotropic. Since point charges are
by definition spherical symmetric, their interaction is isotropic. Especially in the
older literature, intermolecular interactions are regularly assumed to be
isotropic (e.g., the potential is described in


