To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Multi-configurational self-consistent field
Multi-configurational self-consistent field (MCSCF) is a method in quantum chemistry used to generate qualitatively correct reference states of molecules in cases where Hartree-Fock and density functional theory are not adequate (e.g., for molecular ground states which are quasi-degenerate with low lying excited states or in bond breaking situations). It uses a linear combination of configuration state functions (CSF) or configuration determinants to approximate the exact electronic wavefunction of an atom or molecule. In an MCSCF calculation, the set of coefficients of both the CSFs or determinants and the basis functions in the molecular orbitals are varied to obtain the total electronic wavefunction with the lowest possible energy. This method can be considered an intermediate between configuration interaction (where the molecular orbitals are not varied but the expansion of the wave function) and Hartree-Fock (where there is only one determinant but the molecular orbitals are varied). MCSCF wave functions are often used as reference states for Multireference configuration interaction (MRCI) or multi-reference perturbation theories like Complete Active Space Perturbation Theory (CASPT2). These methods can deal with extremely complex chemical situations and, if computing power permits, may be used to reliably calculate molecular ground- and excited states if all other methods fail. Additional recommended knowledge
IntroductionFor the simplest single bond, found in the H2 molecule, molecular orbitals can always be written in terms of two functions χiA and χiB (which are atomic orbitals with small corrections) located at the two nuclei,
where Ni is a normalization constant. The ground state wavefunction for H2 at the equilibrium geometry is dominated by the configuration (φ1)2, which means the molecular orbital φ1 is nearly doubly occupied. The Hartree-Fock model assumes it is doubly occupied, which leads to a total wavefunction of
where Θ2,0 is the singlet (S=0) spin function for two electrons. The molecular orbitals in this case φ1 are taken as sums of 1s atomic orbitals on both atoms, namely N1(1sA+1sB). Expanding the above equation into atomic orbitals yields
This Hartree-Fock model gives a reasonable description of H2 around the equilibrium geometry - about 0.735Å for the bond length (compared to a 0.746Å experimental value) and 84 kcal/mol for the bond energy (exp. 109 kcal/mol). This is typical of the HF model, which usually describes closed shell systems around their equilibrium geometry quite well. At large separations, however, the terms describing both electrons located at one atom remain, which corresponds to dissociation to H+ + H−, which has a much larger energy than H + H. Therefore, the persisting presence of ionic terms leads to an unphysical solution in this case. Consequently, the HF model cannot be used to describe dissociation processes with open shell products. The most straightforward solution to this problem is introducing coefficients in front of the different terms in Ψ1:
which forms the basis for the valence bond description of chemical bonds. With the coefficients CIon and CCov varying, the wave function will have the correct form, with CIon=0 for the separated limit and CIon comparable to CCov at equilibrium. Such a description, however, uses non-orthogonal basis functions, which complicates its mathematical structure. Instead, multiconfiguration is achieved by using orthogonal molecular orbitals. After introducing an anti-bonding orbital
the total wave function of H2 can be written as a linear combination of configurations built from bonding and anti-bonding orbitals:
where Φ2 is the electronic configuration (φ2)2. In this multiconfigurational description of the H2 chemical bond, C1=1 and C2=0 close to equilibrium, and C1 will be comparable to C2 for large separations.[1] Complete Active Space SCFA particularly important MCSCF approach is the complete active space SCF method (CASSCF), where the linear combination of CSFs includes all that arise from a particular number of electrons in a particular number of orbitals. For example, one might define CASSCF(11,8) for the molecule, NO, where the 11 valence electrons are distributed between all configurations that can be constructed from 8 molecular orbitals.[2] [3] Restricted Active Space SCFSince the number of CSFs quickly increases with the number of active orbitals, along with the computational cost, it may be desirable to use a smaller set of CSFs. One way to make this selection is to restrict the number of electrons in certain subspaces, done in the restricted active space SCF method (RASSCF). One could, for instance, allow only single and double excitations from some strongly-occupied subset of active orbitals, or restrict the number of electrons to at most 2 in another subset of active orbitals. See also
References
Further reading
Categories: Quantum chemistry | Computational chemistry |
||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Multi-configurational_self-consistent_field". A list of authors is available in Wikipedia. |



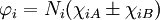 ,
,
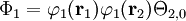 ,
,
![\Phi_1 = N_1^2 \left[ 1s_A(\mathbf{r}_1)1s_A(\mathbf{r}_2) + 1s_A(\mathbf{r}_1)1s_B(\mathbf{r}_2) + 1s_B(\mathbf{r}_1)1s_A(\mathbf{r}_2) + 1s_B(\mathbf{r}_1)1s_B(\mathbf{r}_2) \right] \Theta_{2,0}](images/math/e/4/3/e43c56782e4c67eb533d537bfc2cd6e2.png) .
.


