To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
PharmacodynamicsPharmacodynamics is the study of the biochemical and physiological effects of drugs on the body or on microorganisms or parasites within or on the body and the mechanisms of drug action and the relationship between drug concentration and effect.[1] One dominant example being drug-receptor interactions as modeled by: where L=ligand (drug), R=receptor (attachment site), reaction dynamics that can be studied mathematically through tools such as free energy maps. Pharmacodynamics is often summarized as the study of what a drug does to the body, whereas pharmacokinetics is the study of what the body does to a drug. Pharmacodynamics is sometimes abbreviated as "PD", and when referred to in conjunction with pharmacokinetics can be referred to as "PKPD". Additional recommended knowledge
Effects on the bodyThe majority of drugs either (a) mimic or inhibit normal physiological/biochemical processes or inhibit pathological processes in animals or (b) inhibit vital processes of endo- or ectoparasites and microbial organisms. There are 4 main drug actions:
Desired activityThe desired activity of a drug is mainly due to one of the following:
General anesthetics were once thought to work by disordering the neural membranes, thereby altering the Na+ influx. Antacids and chelating agents combine chemically in the body. Enzyme-substrate binding is a way to alter the production or metabolism of key endogenous chemicals, for example aspirin irreversibly inhibits the enzyme prostaglandin synthetase (cyclooxygenase) thereby preventing inflammatory response. Colchicine, a drug for gout, interferes with the function of the structural protein tubulin, while Digitalis, a drug still used in heart failure, inhibits the activity of the carrier molecule, Na-K-ATPase pump. The widest class of drugs act as ligands which bind to receptors which determine cellular effects. Upon drug binding, receptors can elicit their normal action (agonist), blocked action (antagonist), or even action opposite to normal (inverse agonist). In principle, a pharmacologist would aim for a target plasma concentration of the drug for a desired level of response. In reality, there are many factors affecting this goal. Pharmacokinetic factors determine peak concentrations, and concentrations cannot be maintained with absolute consistency because of metabolic breakdown and excretory clearance. Genetic factors may exist which would alter metabolism or drug action itself, and a patient's immediate status may also affect indicated dosage. Undesirable effectsUndesirable effects of a drug include:
Therapeutic windowThe therapeutic window is the amount of a medication between the amount that gives an effect (effective dose) and the amount that gives more adverse effects than desired effects[2]. For instance, medication with a small pharmaceutical window must be administered with care and control, e.g. by frequently measuring blood concentration of the drug, since it easily loses effects or gives adverse effects. Receptor bindingThe binding of ligands (drug) to receptors is governed by the law of mass action which relates the large-scale status to the rate of numerous molecular processes. The rates of formation and un-formation can be used to determine the equilibrium concentration of bound receptors. The equilibrium dissociation constant is defined by: where L=ligand, R=receptor, square brackets [] denote concentration. The fraction of bound receptors is found as (1+[R]/[L·R])-1 , which can then be expressed using Kd as,
This expression is one way to consider the effect of a drug, in which the response is related to the fraction of bound receptors. The fraction of bound receptors is known as occupancy. The relationship between occupancy and pharmacological response is usually non-linear. This explains the so called receptor reserve phenomenon i.e. the concentration producing 50% occupancy is typically higher than the concentration producing 50% of maximum response. Often the response is determined as a function of log[L] to consider many orders of magnitude of concentration. However, there is no biological or physical theory which relates effects to the log of concentration. It is just convenient for graphing purposes. It is useful to note that 50% of the receptors are bound when [L]=Kd . The graph shown represents the conc-response for two hypothetical receptor agonists, plotted in a semi-log fashion. The curve toward the left represents a higher potency (potency arrow does not indicate direction of increase) since lower concentrations are needed for a given response. The effect increases as a function of concentration. Multicellular pharmacodynamicsThe concept of pharmacodynamics has been expanded to include Multicellular Pharmacodynamics (MCPD). MCPD is the study of the static and dynamic properties and relationships between a set of drugs and a dynamic and diverse multicellular 4 dimensional organization. It is the study of the workings of a drug on a minimal multicellular system (mMCS), both in vivo and in silico. Networked Multicellular Pharmacodynamics (Net-MCPD) further extends the concept of MCPD to model regulatory genomic networks together with signal transduction pathways, as part of a complex of interacting components in the cell. For a fuller explanation of these concepts see the articles:
A good source for further information and posting to experts can be found courtesy of Dr. David W. A. Bourne, OU College of Pharmacy [1]. See alsoReferences
Categories: Pharmacodynamics | Pharmacy | Medicinal chemistry |
|||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Pharmacodynamics". A list of authors is available in Wikipedia. |
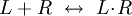



![K_d = \frac{[L][R]}{[L\! \cdot \!R]}](images/math/c/f/b/cfb1b0beb4fdfb3f00a7718e79a8ac82.png)
![Fraction \ Bound = \frac{1}{1+\frac{K_d}{[L]}}](images/math/6/2/e/62e1c65d042c3132e4f1ac972e108279.png)


