Le retour en arrière pour prédire les réactions chimiques à plusieurs étapes
Les chercheurs surmontent les limites du calcul pour prédire les produits de départ de réactions à plusieurs étapes en utilisant uniquement des informations sur la molécule de produit cible.
Vous est-il déjà arrivé de ne voir que la fin d'une émission de télévision et de vous demander comment l'histoire a progressé jusqu'à cette fin ? De la même manière, les chimistes ont souvent une molécule désirée en tête et se demandent quel type de réaction pourrait la produire. Les chercheurs du groupe Maeda de l'Institute for Chemical Reaction Design and Discovery (ICReDD) et de l'université d'Hokkaido ont mis au point une méthode permettant de prédire l'"histoire" (c'est-à-dire les matières premières et les voies de réaction) de réactions chimiques à plusieurs étapes en utilisant uniquement des informations sur la "fin" (c'est-à-dire les molécules produites).
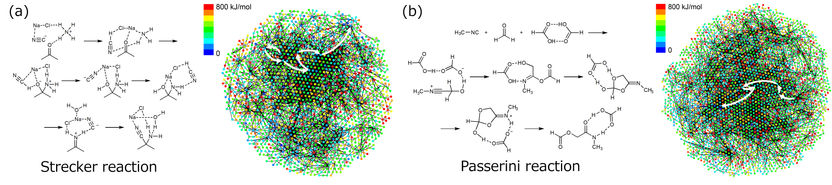
Structures générées et réseaux de chemins de réaction pour les étapes de réaction prédites pour la réaction de Strecker (a) et la réaction de Passerini (b). Les flèches blanches montrent le chemin multi-étapes correspondant au mécanisme de réaction connu.
Satoshi Maeda
Prédire la recette d'une molécule de produit cible, sans autre connaissance que la molécule elle-même, serait un outil puissant pour accélérer la découverte de nouvelles réactions. Le groupe Maeda a déjà mis au point une méthode de calcul qui a permis de prédire les réactions à une seule étape de cette manière. Cependant, l'extension aux réactions à plusieurs étapes entraîne une augmentation spectaculaire du nombre de voies de réaction possibles - ce que l'on appelle une explosion combinatoire. Cette forte augmentation de la complexité entraîne des coûts de calcul prohibitifs.
Pour surmonter cette limitation, les chercheurs ont mis au point un algorithme qui réduit le nombre de chemins à explorer en écartant les chemins moins viables à chaque étape de la réaction. Après avoir calculé tous les chemins possibles pour une étape en arrière dans la réaction, une méthode d'analyse cinétique évalue dans quelle mesure chaque chemin produit la molécule cible. Les voies de réaction qui ne produisent pas la molécule cible au-delà d'un pourcentage seuil préétabli sont considérées comme non significatives et ne sont pas explorées plus avant.
Ce cycle d'exploration, d'évaluation et d'élimination des voies de réaction est répété pour chaque étape en arrière dans une réaction à plusieurs étapes et atténue l'explosion combinatoire qui se produirait normalement, ce qui rend les réactions à plusieurs étapes plus faciles à calculer. Les méthodes précédentes étaient limitées aux réactions à une seule étape, alors que cette nouvelle méthode a pu prédire des réactions comportant plus de 6 étapes, marquant ainsi un saut important dans les capacités.
Comme preuve de concept, les chercheurs ont testé la méthode sur deux réactions multi-étapes bien connues, les réactions de Strecker et de Passerini. Des milliers de produits de départ candidats ont été proposés pour chaque réaction, qui ont été filtrés pour en retenir les plus prometteurs en fonction de leur stabilité et du rendement du produit. Parmi les candidats proposés figuraient les produits de départ bien connus pour chaque réaction, ce qui confirme la capacité de la technique à identifier des produits de départ expérimentalement viables à partir de la seule molécule du produit cible.
Bien que des travaux supplémentaires soient nécessaires pour permettre la prédiction de systèmes encore plus grands et plus complexes, les chercheurs prévoient que cette avancée dans le traitement des processus à étapes multiples accélérera la découverte de nouvelles réactions chimiques.
"Ce travail fournit une approche unique, car c'est la première fois qu'il est possible de réaliser des prédictions inverses de réactions à plusieurs étapes en utilisant des calculs de chimie quantique sans utiliser aucune connaissance ou donnée sur la réaction", a déclaré le professeur Satoshi Maeda. "Nous pensons que cette technique permettra de découvrir des transformations chimiques totalement inimaginables, dans lesquelles il n'y a que peu de connaissances ou de données expérimentales à utiliser."
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale
Autres actualités du département science















Recevez les dernières actualités de l’industrie chimique
Ne manquez plus aucune actualité : notre newsletter consacrée à l'industrie chimique, aux méthodes analytiques, aux laboratoires et à la technologie des processus vous informe tous les mardis et jeudis. Les dernières nouvelles du secteur, les produits phares et les innovations - de manière compacte et compréhensible dans votre boîte de réception. Recherché par nos soins, pour que vous n'ayez pas à le faire.