Il potere dei gechi
La TU Wien risolve il puzzle delle grandi molecole
Annunci


Un rompicapo della chimica teorica è stato risolto alla TU Wien: un nuovo metodo di calcolo consente ora di calcolare le forze tra grandi molecole con una precisione senza precedenti.
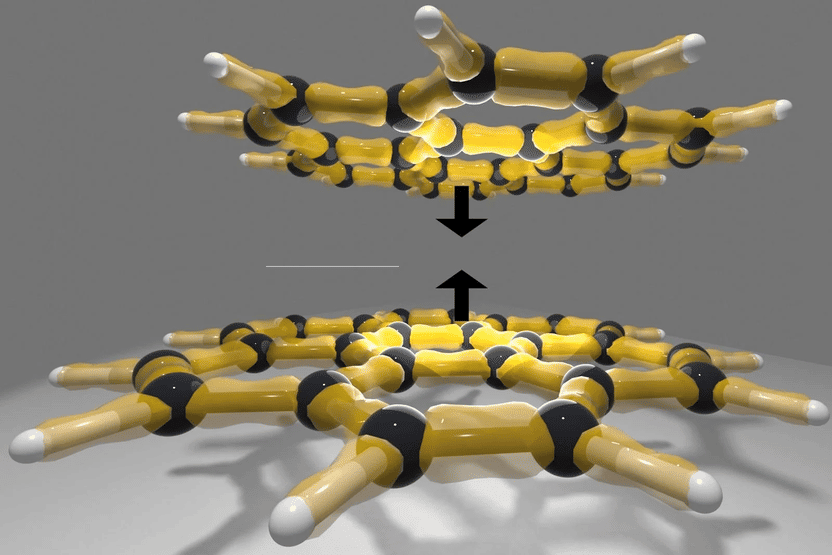
Coronene-Dimero: un esempio di grandi molecole con forze di Van-der-Waals
© TU Wien
Perché i gechi possono camminare sui muri? Perché l'azoto diventa liquido a -196 °C? Molti fenomeni quotidiani possono essere spiegati dalle forze di van der Waals, legami deboli tra molecole che sono notoriamente difficili da calcolare. Per anni gli scienziati hanno dovuto fare i conti con il fatto che diversi metodi di calcolo producevano risultati contrastanti.
Ora i ricercatori della TU Wien hanno risolto questa discrepanza e trovato una soluzione. Ironia della sorte, proprio il metodo a lungo considerato il "gold standard" della chimica quantistica si è rivelato la fonte dell'errore: sovrastima sistematicamente l'energia contenuta in alcuni legami molecolari. Con una variante migliorata, il team della TU Wien è ora in grado di prevedere correttamente il comportamento di grandi molecole, un passo essenziale per la comprensione dei sistemi biologici e per il progresso delle tecnologie delle energie rinnovabili.
Un mistero della chimica
"Per descrivere i legami tra grandi molecole, gli scienziati utilizzano diversi approcci computazionali", spiegano Tobias Schäfer e Andreas Irmler, primi autori del nuovo studio. Insieme ad Alejandro Gallo e al leader del gruppo di ricerca, il Prof. Andreas Grüneis, hanno confrontato i metodi più utilizzati.
"Una possibilità è quella di utilizzare simulazioni quantistiche Monte Carlo", spiega Schäfer. "In questo caso, il computer esplora innumerevoli possibili disposizioni di elettroni, mantenendo quelle energeticamente favorevoli e scartando quelle sfavorevoli. Un'altra opzione è il cosiddetto approccio a cluster accoppiati", aggiunge Irmler. "In questo caso, le molecole vengono trattate nei loro stati a bassa energia e le configurazioni a più alta energia vengono aggiunte in seguito come una sorta di correzione".
"Questo metodo a cluster accoppiati è stato a lungo considerato il gold standard", afferma Schäfer. "Ma più guardavamo da vicino, più diventava chiaro che c'erano piccole ma persistenti deviazioni rispetto ai risultati di Monte Carlo, e per anni nessuno sapeva perché".
Ora il team della TU Wien ha trovato la risposta: "Abbiamo scoperto che il metodo dei cluster accoppiati sovrastima sistematicamente le energie di legame in molecole grandi e altamente polarizzabili", spiega Irmler. "La nostra variante migliorata corregge questa deviazione senza aumentare significativamente il costo computazionale". Con questa correzione, i risultati sono ora molto più vicini ai dati di Monte Carlo quantistico.
Grandi molecole - Grande importanza
Questo progresso è particolarmente importante per i sistemi molecolari di grandi dimensioni. "Se si vogliono descrivere molecole contenenti fino a un centinaio di atomi, lo sforzo computazionale diventa enorme", spiega Alejandro Gallo. "Anche i più grandi supercomputer del mondo raggiungono i loro limiti. Per ottenere previsioni affidabili, abbiamo bisogno di metodi di approssimazione molto sofisticati".
E le molecole di grandi dimensioni stanno diventando sempre più importanti, in campi che vanno dalla ricerca sui materiali allo sviluppo farmaceutico. "Se vogliamo capire come un farmaco si cristallizza all'interno di una compressa o quanto fortemente un materiale lega l'idrogeno per l'immagazzinamento dell'energia, dobbiamo modellare accuratamente le forze di van der Waals", afferma Schäfer.
Dalla teoria fondamentale alle applicazioni pratiche
Il nuovo metodo consente di ottenere dati di riferimento più affidabili, non solo per le simulazioni tradizionali ma anche come dati di addestramento per i modelli di intelligenza artificiale. Tali modelli vengono già utilizzati per progettare nuovi materiali e prodotti farmaceutici in ambienti virtuali.
"Stiamo creando un ponte tra la massima precisione e l'usabilità pratica", afferma il Prof. Andreas Grüneis dell'Istituto di Fisica Teorica della TU Wien. "Questo apre nuove possibilità per la scienza dei materiali. I nostri risultati dimostrano che anche i metodi consolidati devono essere continuamente riesaminati per tenere il passo con le crescenti esigenze della ricerca moderna".
Nota: questo articolo è stato tradotto utilizzando un sistema informatico senza intervento umano. LUMITOS offre queste traduzioni automatiche per presentare una gamma più ampia di notizie attuali. Poiché questo articolo è stato tradotto con traduzione automatica, è possibile che contenga errori di vocabolario, sintassi o grammatica. L'articolo originale in Inglese può essere trovato qui.

Altre notizie dal dipartimento scienza














































