Un nuovo metodo migliora l'accuratezza dei potenziali appresi dalla macchina per la simulazione dei catalizzatori
Un ponte tra l'accuratezza e l'efficienza nella progettazione dei catalizzatori
Annunci



I catalizzatori svolgono un ruolo indispensabile nella produzione moderna. Oltre l'80% di tutti i prodotti manifatturieri, dai farmaci alle materie plastiche, si basa su processi catalitici in qualche fase della produzione. I metalli di transizione, in particolare, si distinguono come catalizzatori molto efficaci perché i loro orbitali d parzialmente riempiti consentono loro di scambiare facilmente elettroni con altre molecole. Proprio questa proprietà, tuttavia, li rende difficili da modellare con precisione, richiedendo descrizioni precise della loro struttura elettronica.
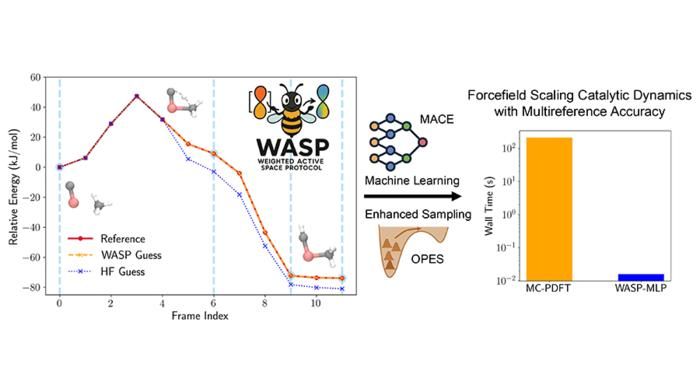
Precisione e velocità raggiunte con il protocollo WASP (Weighted Active Space Protocol) per l'attivazione del metano sul carburo di titanio.
Figure courtesy of Seal et al.
La progettazione di catalizzatori efficienti a base di metalli di transizione, in grado di funzionare in condizioni realistiche, richiede più di un'istantanea statica di una reazione. Dobbiamo invece catturare il quadro dinamico: le molecole si muovono e interagiscono a temperature e pressioni diverse, dove il movimento atomico modella fondamentalmente le prestazioni catalitiche.
Per affrontare questa sfida, il laboratorio della professoressa Laura Gagliardi della Pritzker School of Molecular Engineering (UChicago PME) e del Dipartimento di Chimica dell'Università di Chicago ha sviluppato un nuovo potente strumento che sfrutta le teorie della struttura elettronica e l'apprendimento automatico per simulare la dinamica catalitica dei metalli di transizione con precisione e velocità.

"Nell'ultimo decennio, i potenziali appresi dalla macchina hanno fatto progredire in modo significativo il modo in cui simuliamo la dinamica molecolare, offrendo velocità e scalabilità. Tuttavia, la cattura accurata della struttura elettronica dei catalizzatori di metalli di transizione è rimasta una sfida irrisolta. Il nostro nuovo metodo colma questa lacuna integrando metodi di chimica quantistica multireferenziale con potenziali machine learned, garantendo precisione ed efficienza". ha dichiarato Gagliardi.
I risultati sono stati pubblicati nei Proceedings of the National Academy of Sciences.
Consentire l'apprendimento automatico per accelerare le simulazioni
Negli ultimi dieci anni, il gruppo di Gagliardi ha sviluppato la teoria funzionale della densità di coppia multiconfigurazione (MC-PDFT), un metodo di chimica quantistica in grado di descrivere le intricate strutture elettroniche delle reazioni dei metalli di transizione. Sebbene la MC-PDFT offra un'elevata precisione, è proibitivamente lenta per simulare la dinamica dei sistemi catalitici, un passo fondamentale per prevedere il comportamento reale dei catalizzatori in condizioni realistiche.
Per affrontare questa sfida, il team si è rivolto ai potenziali interatomici appresi dalla macchina (ML-potenziali), che possono catturare la dinamica molecolare con notevole efficienza. I potenziali ML sono stati ampiamente applicati nella scienza dei materiali, ma finora non erano mai stati combinati con successo con metodi multireferenza come MC-PDFT.
Il motivo risiede in un ostacolo di vecchia data: la coerenza dell'etichettatura. I modelli di apprendimento automatico richiedono etichette di proprietà uniche e affidabili, come le energie e le forze derivate dalle funzioni d'onda, per ogni geometria molecolare lungo un percorso di reazione. Per i metodi di chimica quantistica multireferenza, l'assegnazione di tali etichette in modo univoco è rimasta un problema irrisolto.
Per superare questa sfida, lo studente di dottorato Aniruddha Seal, con la consulenza congiunta di Gagliardi e del professor Andrew Ferguson, ha sviluppato un nuovo algoritmo che genera funzioni d'onda coerenti per le nuove geometrie come combinazione ponderata di funzioni d'onda di strutture molecolari precedentemente campionate. Più una nuova geometria è vicina a una conosciuta, più la sua funzione d'onda assomiglia a quella della struttura conosciuta. Questo approccio garantisce che a ogni punto del percorso di reazione venga assegnata una funzione d'onda unica e coerente, consentendo di addestrare accuratamente i potenziali ML su dati multireferenza.
"Pensate a come mescolare i colori su una tavolozza", ha spiegato Seal. "Se voglio creare una tonalità di verde più vicina al blu, userò più vernice blu e un po' di giallo. Se voglio una tonalità che tende al giallo, l'equilibrio si inverte. Più il mio colore target è vicino a una delle vernici di base, più influenza la miscela. Il WASP funziona allo stesso modo: fonde le informazioni provenienti dalle strutture molecolari vicine, dando maggior peso a quelle più simili, per creare una previsione accurata della nuova geometria".
Questa innovazione è alla base del Weighted Active Space Protocol (WASP), una struttura che combina l'accuratezza della MC-PDFT con l'efficienza dell'apprendimento automatico, sviluppata grazie alla stretta collaborazione con il gruppo Parrinello dell'Istituto Italiano di Tecnologia di Genova, che riunisce le competenze della teoria della struttura elettronica e dei potenziali appresi dalla macchina. WASP consente di ottenere accelerazioni notevoli: le simulazioni con accuratezza multireferenziale che un tempo richiedevano mesi possono ora essere completate in pochi minuti.
Impatto: Un ponte tra precisione ed efficienza nella progettazione dei catalizzatori
Unendo precisione e velocità, WASP apre le porte alla progettazione di catalizzatori in grado di resistere a condizioni realistiche: alte temperature e alte pressioni. I metalli di transizione sono fondamentali per innumerevoli processi su larga scala, ma la loro complessità ha reso difficile la progettazione razionale dei catalizzatori.
Un esempio lampante è il processo Haber-Bosch, in cui il ferro funge da catalizzatore per convertire azoto e idrogeno in ammoniaca. Nonostante sia stato sviluppato più di un secolo fa, questo catalizzatore di ferro domina ancora la produzione di ammoniaca in tutto il mondo. Con WASP, i ricercatori hanno ora gli strumenti per esplorare alternative che potrebbero aumentare l'efficienza, ridurre i sottoprodotti e diminuire il costo ambientale.
Finora il WASP è stato dimostrato con successo per la catalisi attivata termicamente, ossia per le reazioni guidate dal calore. La prossima frontiera è l'adattamento del metodo alle reazioni attivate dalla luce, che sono essenziali per la progettazione di nuovi fotocatalizzatori. I fotocatalizzatori sono molto promettenti per le tecnologie, dal trattamento delle acque alla produzione di energia.
Nota: questo articolo è stato tradotto utilizzando un sistema informatico senza intervento umano. LUMITOS offre queste traduzioni automatiche per presentare una gamma più ampia di notizie attuali. Poiché questo articolo è stato tradotto con traduzione automatica, è possibile che contenga errori di vocabolario, sintassi o grammatica. L'articolo originale in Inglese può essere trovato qui.
Pubblicazione originale




Altre notizie dal dipartimento scienza










































