Novo método melhora a precisão dos potenciais aprendidos por máquina para simular catalisadores
Estabelecer uma ponte entre precisão e eficiência na conceção de catalisadores
Anúncios



Os catalisadores desempenham um papel indispensável no fabrico moderno. Mais de 80% de todos os produtos manufacturados, desde os produtos farmacêuticos aos plásticos, dependem de processos catalíticos em alguma fase da produção. Os metais de transição, em particular, destacam-se como catalisadores altamente eficazes porque as suas orbitais d parcialmente preenchidas lhes permitem trocar facilmente electrões com outras moléculas. No entanto, esta propriedade torna difícil modelá-los com precisão, exigindo descrições precisas da sua estrutura eletrónica.
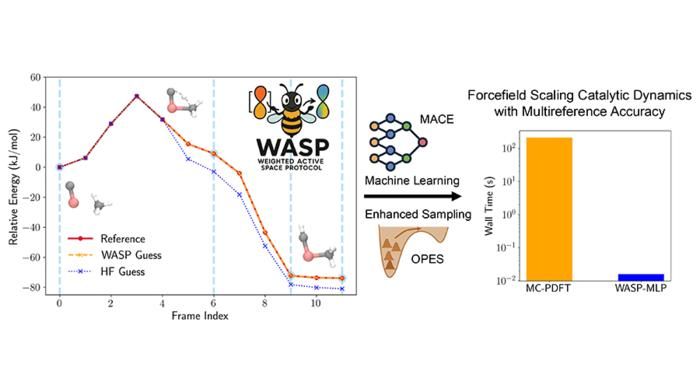
Precisão e aumento de velocidade obtidos com o Protocolo de Espaço Ativo Ponderado (WASP) para a ativação de metano em carboneto de titânio.
Figure courtesy of Seal et al.
A conceção de catalisadores eficientes de metais de transição que possam funcionar em condições realistas exige mais do que uma fotografia estática de uma reação. Em vez disso, temos de captar o quadro dinâmico - como as moléculas se movem e interagem a diferentes temperaturas e pressões, onde o movimento atómico molda fundamentalmente o desempenho catalítico.
Laura Gagliardi, da Escola Pritzker de Engenharia Molecular da Universidade de Chicago (UChicago PME) e do Departamento de Química, desenvolveu uma nova e poderosa ferramenta que utiliza teorias de estrutura eletrónica e aprendizagem automática para simular a dinâmica catalítica de metais de transição com precisão e rapidez.

"Na última década, os potenciais aprendidos por máquinas fizeram avançar significativamente a forma como simulamos a dinâmica molecular, oferecendo velocidade e escalabilidade. No entanto, capturar com precisão a estrutura eletrónica dos catalisadores de metais de transição continua a ser um desafio por resolver. O nosso novo método preenche esta lacuna, integrando métodos de química quântica multireferencial com potenciais aprendidos por máquina, proporcionando precisão e eficiência". afirmou Gagliardi.
Os resultados foram publicados na revista Proceedings of the National Academy of Sciences.
Permitir a aprendizagem automática para acelerar as simulações
Na última década, o grupo de Gagliardi desenvolveu a teoria funcional de densidade de pares multiconfiguração (MC-PDFT), um método de química quântica capaz de descrever as intrincadas estruturas electrónicas das reacções dos metais de transição. Embora a MC-PDFT ofereça uma elevada precisão, é proibitivamente lenta para simular a dinâmica dos sistemas catalíticos - um passo crítico na previsão do verdadeiro comportamento dos catalisadores em condições realistas.
Para enfrentar este desafio, a equipa recorreu a potenciais interatómicos aprendidos por máquina (potenciais ML), que podem captar a dinâmica molecular com uma eficiência notável. Os potenciais ML têm sido amplamente aplicados na ciência dos materiais, mas até agora nunca tinham sido combinados com sucesso com métodos de multireferência como o MC-PDFT.
A razão reside num obstáculo de longa data: a consistência da rotulagem. Os modelos de aprendizagem automática requerem rótulos de propriedades únicos e fiáveis - tais como energias e forças derivadas de funções de onda - para cada geometria molecular ao longo de um percurso de reação. Para os métodos de química quântica multireferencial, a atribuição de tais rótulos de forma única permaneceu um problema não resolvido.
Andrew Ferguson, desenvolveu um novo algoritmo que gera funções de onda consistentes para novas geometrias como uma combinação ponderada de funções de onda de estruturas moleculares previamente amostradas. Quanto mais próxima uma nova geometria estiver de uma conhecida, mais fortemente a sua função de onda se assemelha à da estrutura conhecida. Esta abordagem assegura que a cada ponto ao longo de uma via de reação é atribuída uma função de onda única e consistente, tornando possível treinar potenciais ML com precisão em dados multi-referência.
"Pense nisso como misturar tintas numa paleta", explicou Seal. "Se eu quiser criar um tom de verde mais próximo do azul, utilizo mais tinta azul e apenas um pouco de amarelo. Se quiser um tom mais próximo do amarelo, o equilíbrio inverte-se. Quanto mais próxima a minha cor alvo estiver de uma das tintas de base, mais influencia a mistura. O WASP funciona da mesma forma: mistura informações de estruturas moleculares próximas, dando mais peso às que são mais semelhantes, para criar uma previsão exacta da nova geometria."
Esta inovação constitui a base do Protocolo de Espaço Ativo Ponderado (WASP), uma estrutura que combina a precisão do MC-PDFT com a eficiência da aprendizagem automática, desenvolvida através de uma estreita colaboração com o Grupo Parrinello do Instituto Italiano de Tecnologia de Génova, reunindo conhecimentos especializados em teoria da estrutura eletrónica e potenciais aprendidos por máquina. O WASP permite aumentos drásticos de velocidade: simulações com precisão multireferencial que antes demoravam meses podem agora ser concluídas em apenas alguns minutos.
Impacto: Ligar a precisão e a eficiência na conceção de catalisadores
Ao unir precisão e velocidade, o WASP abre a porta para a conceção de catalisadores que podem suportar condições realistas - altas temperaturas e altas pressões. Os metais de transição são fundamentais para inúmeros processos em grande escala, mas a sua complexidade tornou a conceção racional de catalisadores um desafio.
Um bom exemplo é o processo Haber-Bosch, em que o ferro serve de catalisador para converter azoto e hidrogénio em amoníaco. Apesar de ter sido desenvolvido há mais de um século, este catalisador de ferro continua a dominar a produção de amoníaco em todo o mundo. Com o WASP, os investigadores dispõem agora de ferramentas para explorar alternativas que podem aumentar a eficiência, reduzir os subprodutos e diminuir o custo ambiental.
Até agora, o WASP foi demonstrado com sucesso para a catálise activada termicamente - reacções impulsionadas pelo calor. A próxima fronteira é a adaptação do método a reacções activadas pela luz, que são essenciais para a conceção de novos fotocatalisadores. Os fotocatalisadores são uma enorme promessa para as tecnologias, desde o tratamento da água até à produção de energia.
Observação: Este artigo foi traduzido usando um sistema de computador sem intervenção humana. A LUMITOS oferece essas traduções automáticas para apresentar uma gama mais ampla de notícias atuais. Como este artigo foi traduzido com tradução automática, é possível que contenha erros de vocabulário, sintaxe ou gramática. O artigo original em Inglês pode ser encontrado aqui.
Publicação original




Outras notícias do departamento ciência














































