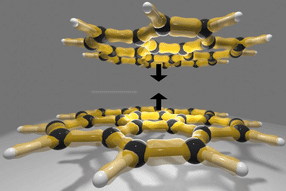
Previsione della struttura cristallina di molecole organiche più rapida e affidabile

Annunci


La previsione delle strutture cristalline delle molecole organiche è un compito critico in molti settori, soprattutto in quello farmaceutico e nella progettazione di materiali funzionali. Nel settore farmaceutico, le strutture cristalline influenzano direttamente la solubilità e la stabilità di un farmaco. Nei materiali funzionali, come i semiconduttori organici, il controllo delle strutture cristalline è fondamentale per ottenere le proprietà elettroniche desiderate. Tuttavia, la previsione della struttura cristallina (CSP) è un compito intrinsecamente difficile a causa delle deboli e diverse interazioni intra- e inter-molecolari proprie dei cristalli organici. Anche piccole variazioni possono portare a disposizioni di impacchettamento completamente diverse.
La CSP è tipicamente condotta in due fasi: esplorazione della struttura e rilassamento della struttura. Nella prima fase, viene generato un gran numero di strutture potenziali, spesso casuali, per le quali sono stati sviluppati vari algoritmi di ricerca. Durante il rilassamento della struttura, queste strutture vengono raffinate per identificare le configurazioni più stabili utilizzando la minimizzazione dell'energia. Tuttavia, la generazione casuale di strutture spesso produce diverse strutture a bassa densità e instabili, mentre i metodi convenzionali basati sulla teoria funzionale della densità (DFT) per il rilassamento della struttura sono computazionalmente costosi e richiedono molto tempo.
Per affrontare queste sfide, il professore associato Takuya Taniguchi del Center for Data Science e Ryo Fukasawa della Graduate School of Advanced Science and Engineering della Waseda University, in Giappone, hanno sviluppato un flusso di lavoro CSP basato sull'apprendimento automatico (ML) chiamato SPaDe-CSP che sfrutta i predittori del gruppo spaziale (SP) e della densità di impacchettamento (PD). "Il nostro flusso di lavoro impiega una strategia unica in cui i modelli di apprendimento automatico prevedono innanzitutto i gruppi spaziali e le densità cristalline più probabili, filtrando i candidati instabili e a bassa densità prima delle fasi di rilassamento computazionalmente intensive", spiega Taniguchi. Insieme a un potenziale di rete neurale efficiente per il rilassamento della struttura, questo metodo consente un percorso più diretto e affidabile per identificare le disposizioni cristalline osservate sperimentalmente". "Il loro studio è stato pubblicato sulla rivista Digital Discovery il 13 ottobre 2025.

SPaDe-CSP restringe lo spazio di ricerca dei cristalli organici, prevedendo innanzitutto i probabili gruppi spaziali candidati e le densità cristalline mediante modelli ML. Per l'addestramento e il test, i ricercatori hanno estratto un set di dati dal Cambridge Structural Database (CSD), composto da 32 candidati gruppi spaziali con 169.656 voci di dati. Entrambi i modelli di previsione hanno utilizzato MACCSKeys come impronta molecolare e LightGBM come funzione di previsione. I ricercatori hanno anche interpretato i modelli addestrati utilizzando l'analisi delle spiegazioni additive di Shapley (SHAP) per identificare le caratteristiche strutturali più importanti per una previsione efficace.
Dopo il campionamento del reticolo, le strutture non rilassate generate vengono sottoposte al rilassamento della struttura utilizzando un potenziale di rete neurale efficiente (NNP) preaddestrato sui dati DFT, producendo infine il diagramma di densità energetica della molecola target. Due iperparametri controllano il processo SPaDe-CSP: la soglia di probabilità per il filtraggio dei gruppi spaziali e la finestra di tolleranza per la densità cristallina.
I ricercatori hanno testato il flusso di lavoro prima su una molecola modello del set di dati CSD per studiare la dipendenza del tasso di successo dagli iperparametri e poi su 20 diverse molecole organiche, compresa la molecola modello, per verificare la generalizzabilità. I risultati sono stati convalidati con successo rispetto alle strutture cristalline sperimentali note delle molecole e confrontati con i risultati ottenuti con il random-CSP convenzionale.
I risultati hanno rivelato che la probabilità di successo aumenta con una soglia di gruppo spaziale più alta e una finestra di tolleranza di densità più piccola. Per l'80% dei composti testati, SPaDe-CSP ha predetto con successo le strutture cristalline sperimentali, ottenendo un tasso di successo doppio rispetto a random-CSP. In particolare, i ricercatori hanno anche identificato un descrittore strutturale chiave che correla linearmente con il tasso di successo, indicando influenze strutturali sia a livello di cristallo che di molecola.
"La nostra strategia può accelerare in modo significativo la progettazione e la scoperta di nuove molecole nell'industria farmaceutica e della scienza dei materiali", afferma Taniguchi. "Ciò consentirà di identificare in modo più rapido e affidabile la forma fisica più stabile ed efficace di un nuovo farmaco, importante per mantenere la solubilità, la durata di conservazione e l'efficacia complessiva, e permetterà lo screening computazionale di nuovi materiali funzionali con proprietà elettroniche ottimali".
Rendendo la CSP più veloce e affidabile, questa ricerca segna un passo importante verso l'accelerazione della scoperta di farmaci salvavita e di tecnologie di nuova generazione.
Nota: questo articolo è stato tradotto utilizzando un sistema informatico senza intervento umano. LUMITOS offre queste traduzioni automatiche per presentare una gamma più ampia di notizie attuali. Poiché questo articolo è stato tradotto con traduzione automatica, è possibile che contenga errori di vocabolario, sintassi o grammatica. L'articolo originale in Inglese può essere trovato qui.
Pubblicazione originale

Altre notizie dal dipartimento scienza