L'intelligence artificielle aide à explorer les frontières de la chimie
L'apprentissage automatique aide à simuler la dynamique moléculaire réactive pour la recherche et la découverte
Annonces



La possibilité de simuler le comportement des systèmes au niveau atomique représente un outil puissant pour tous les domaines, de la conception de médicaments à la découverte de matériaux. Une équipe dirigée par des chercheurs du Los Alamos National Laboratory a mis au point des potentiels interatomiques d'apprentissage automatique qui prédisent les énergies moléculaires et les forces agissant sur les atomes, ce qui permet des simulations plus rapides et moins coûteuses que les méthodes de calcul existantes.
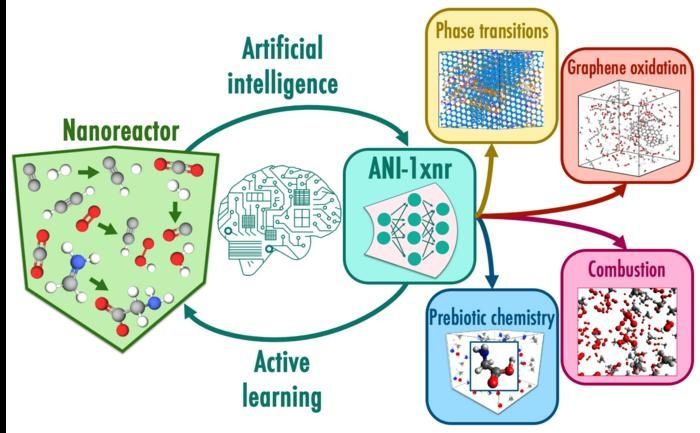
Dans ce flux de travail, les simulations de nanoréacteurs échantillonnent automatiquement l'espace chimique réactif sans dépendre de l'intuition humaine. Le nanoréacteur est une classe spéciale de simulations atomistiques dans lesquelles les réactions chimiques sont induites par la collision de molécules à des vitesses élevées. L'apprentissage actif utilise le potentiel d'apprentissage automatique, ANI-1xnr, pour piloter la dynamique du nanoréacteur et sous-sélectionner les structures présentant des incertitudes élevées. Des études de cas telles que les transitions de phase du carbone et la combustion du méthane testent la généralité du modèle résultant, ANI-1xnr.
Los Alamos National Laboratory
"Les potentiels d'apprentissage automatique offrent de plus en plus une alternative efficace aux simulations coûteuses en calcul qui tentent de représenter des systèmes physiques complexes à l'échelle atomique", a déclaré Benjamin Nebgen, physicien chimiste à Los Alamos et coauteur d'un récent article de Nature Chemistry décrivant les travaux. "Un potentiel interatomique réactif général d'apprentissage automatique, applicable à une large gamme de chimie réactive sans nécessiter de réajustement, profitera grandement à la chimie et à la science des matériaux.
Combler le fossé des simulations efficaces
La construction de simulations efficaces pour la dynamique moléculaire en chimie se fait traditionnellement à l'aide de modèles de calcul basés sur la physique, notamment les champs de force classiques ou la mécanique quantique. Si les modèles de mécanique quantique sont précis et généralement applicables, ils sont extrêmement coûteux en termes de calcul. En revanche, les champs de force classiques sont efficaces en termes de calcul, mais leur précision est relativement faible et ils ne s'appliquent qu'à une gamme limitée de systèmes. ANI-1xnr, le modèle transformationnel d'apprentissage automatique de l'équipe, comble le fossé en termes de vitesse, de précision et de généralité qui existe en chimie depuis plusieurs décennies. (L'apprentissage automatique est une application de l'intelligence artificielle dans laquelle les programmes informatiques "apprennent" par la formation).
ANI-1xnr représente le premier potentiel interatomique réactif d'apprentissage automatique suffisamment général - il peut être appliqué à de nombreux systèmes chimiques différents - pour rivaliser avec les modèles informatiques basés sur la physique pour la réalisation de simulations atomistiques réactives à grande échelle. ANI-1xnr a été développé à l'aide d'un flux de travail automatisé qui a effectué des simulations de dynamique moléculaire réactive sur un large éventail de systèmes chimiques contenant des éléments de carbone, d'hydrogène, d'azote et d'oxygène.
ANI-1xnr s'est avéré capable d'étudier une gamme variée de systèmes, allant des transitions de phase du carbone à la combustion en passant par la chimie prébiotique. L'équipe a validé les simulations en les comparant aux résultats des expériences et des techniques de calcul conventionnelles.
Un potentiel interatomique transformationnel
"ANI-1xnr ne nécessite pas d'expertise dans le domaine ou de réajustement pour chaque nouveau cas d'utilisation, ce qui permet aux scientifiques de divers domaines d'étudier la chimie inconnue", a déclaré Richard Messerly, chercheur en informatique à Los Alamos et coauteur correspondant de l'article. "L'applicabilité générale d'ANI-1xnr est transformationnelle et représente une étape importante vers le remplacement des techniques de modélisation de longue date pour l'étude de la chimie réactive à l'échelle.
L'ensemble des données utilisées par l'équipe et le code ANI-1xnr ont été mis à la disposition de la communauté des chercheurs.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale
Shuhao Zhang, Małgorzata Z. Makoś, Ryan B. Jadrich, Elfi Kraka, Kipton Barros, Benjamin T. Nebgen, Sergei Tretiak, Olexandr Isayev, Nicholas Lubbers, Richard A. Messerly, Justin S. Smith; "Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential"; Nature Chemistry, 2024-3-7




Autres actualités du département recherche et développement















































