This page describes mining for molecules. Since molecules may be represented by molecular graphs this is strongly related to graph mining and structured data mining. The main problem is how to represent molecules while discriminating the data instances. One way to do this is chemical similarity metrics, which has a long tradition in the field of cheminformatics.
Typical approaches to calculate chemical similarities use chemical fingerprints, but this loses the underlying information about the molecule topology. Mining the molecular graphs directly
avoids this problem. So does the inverse QSAR problem which is preferable for vectorial mappings.
Coding(Moleculei,Moleculej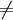 i) i)
Kernel methods
- Marginalized graph kernel[1]
- Optimal assignment kernel[2][3][4]
- Pharmacophore kernel[5]
Maximum Common Graph methods
- MCS-HSCS[6] (Highest Scoring Common Substructure (HSCS) ranking strategy for single MCS)
Coding(Moleculei)
Molecular query methods
See also
References
- Schölkopf, B., K. Tsuda and J. P. Vert: Kernel Methods in Computational Biology, MIT Press, Cambridge, MA, 2004.
- R.O. Duda, P.E. Hart, D.G. Stork, Pattern Classification, John Wiley & Sons, 2001. ISBN 0-471-05669-3
- Gusfield, D., Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology, Cambridge University Press, 1997. ISBN 0-521-58519-8
- R. Todeschini, V. Consonni, Handbook of Molecular Descriptors, Wiley-VCH, 2000. ISBN 3527299130
- ^ H. Kashima, K. Tsuda, A. Inokuchi, Marginalized Kernels Between Labeled Graphs, The 20th International Conference on Machine Learning (ICML2003), 2003. PDF
- ^ H. Fröhlich, J. K. Wegner, A. Zell, Optimal Assignment Kernels For Attributed Molecular Graphs, The 22nd International Conference on Machine Learning (ICML 2005), Omnipress, Madison, WI, USA, 2005, 225-232. PDF
- ^ H. Fröhlich, J. K. Wegner, A. Zell, Kernel Functions for Attributed Molecular Graphs - A New Similarity Based Approach To ADME Prediction in Classification and Regression, QSAR Comb. Sci., 2006, 25, 317-326. DOI 10.1002/qsar.200510135
- ^ H. Fröhlich, J. K. Wegner, A. Zell, Assignment Kernels For Chemical Compounds, International Joint Conference on Neural Networks 2005 (IJCNN'05), 2005, 913-918. CiteSeer
- ^ P. Mahe, L. Ralaivola, V. Stoven, J. Vert, The pharmacophore kernel for virtual screening with support vector machines, J Chem Inf Model, 2006, 46, 2003-2014. DOI 10.1021/ci060138m
- ^ J. K. Wegner, H. Fröhlich, H. Mielenz, A. Zell, Data and Graph Mining in Chemical Space for ADME and Activity Data Sets, QSAR Comb. Sci., 2006, 25, 205-220. DOI 10.1002/qsar.200510009
- ^ T. Meinl, C. Borgelt, M. R. Berthold, Discriminative Closed Fragment Mining and Pefect Extensions in MoFa, Proceedings of the Second Starting AI Researchers Symposium (STAIRS 2004), 2004.
- ^ T. Meinl, C. Borgelt, M. R. Berthold, M. Philippsen, Mining Fragments with Fuzzy Chains in Molecular Databases, Second International Workshop on Mining Graphs, Trees and Sequences (MGTS2004), 2004.
- ^ T. Meinl, M. R. Berthold, Hybrid Fragment Mining with MoFa and FSG, Proceedings of the 2004 IEEE Conference on Systems, Man & Cybernetics (SMC2004), 2004.
- ^ M. Wörlein, Extension and parallelization of a graph-mining-algorithm, Friedrich-Alexander-Universität, 2006. PDF
- ^ A. Clare, R. D. King, Data mining the yeast genome in a lazy functional language, Practical Aspects of Declarative Languages (PADL2003), 2003.
- ^ A. Karwath, L. D. Raedt, SMIREP: predicting chemical activity from SMILES, J Chem Inf Model, 2006, 46, 2432-2444. DOI 10.1021/ci060159g
- ^ R. D. King, A. Srinivasan, L. Dehaspe, Wamr: a data mining tool for chemical data, J. Comput.-Aid. Mol. Des., 2001, 15, 173-181. DOI 10.1023/A:1008171016861
- ^ L. Dehaspe, H. Toivonen, King, Finding frequent substructures in chemical compounds, 4th International Conference on Knowledge Discovery and Data Mining, AAAI Press., 1998, 30-36.
- ^ A. Inokuchi, T. Washio, T. Okada, H. Motoda, Applying the Apriori-based Graph Mining Method to Mutagenesis Data Analysis, Journal of Computer Aided Chemistry, 2001, 2, 87-92.
- ^ A. Inokuchi, T. Washio, K. Nishimura, H. Motoda, A Fast Algorithm for Mining Frequent Connected Subgraphs, IBM Research, Tokyo Research Laboratory, 2002.
- ^ M. Kuramochi, G. Karypis, An Efficient Algorithm for Discovering Frequent Subgraphs, IEEE Transactions on Knowledge and Data Engineering, 2004, 16(9), 1038-1051.
- ^ M. Deshpande, M. Kuramochi, N. Wale, G. Karypis, Frequent Substructure-Based Approaches for Classifying Chemical Compounds, IEEE Transactions on Knowledge and Data Engineering, 2005, 17(8), 1036-1050.
- ^ H. Ando, L. Dehaspe, W. Luyten, E. Craenenbroeck, H. Vandecasteele, L. Meervelt, Discovering H-Bonding Rules in Crystals with Inductive Logic Programming, Mol Pharm, 2006, 3, 665-674 . DOI 10.1021/mp060034z
- ^ S. Nijssen, J. N. Kok. Frequent Graph Mining and its Application to Molecular Databases, Proceedings of the 2004 IEEE Conference on Systems, Man & Cybernetics (SMC2004), 2004.
- ^ K. Jahn, S. Kramer, Optimizing gSpan for Molecular Datasets, Proceedings of the Third International Workshop on Mining Graphs, Trees and Sequences (MGTS-2005), 2005.
- ^ X. Yan, J. Han, gSpan: Graph-Based Substructure Pattern Mining, Proceedings of the 2002 IEEE International Conference on Data Mining (ICDM 2002), IEEE Computer Society, 2002, 721-724.
- ^ C. Helma, T. Cramer, S. Kramer, L. de Raedt, Data Mining and Machine Learning Techniques for the Identification of Mutagenicity Inducing Substructures and Structure Activity Relationships of Noncongeneric Compounds, J. Chem. Inf. Comput. Sci., 2004, 44, 1402-1411. DOI 10.1021/ci034254q
- ^ P. Mazzatorta, L. Tran, B. Schilter, M. Grigorov, Integration of Structure-Activity Relationship and Artificial Intelligence Systems To Improve in Silico Prediction of Ames Test Mutagenicity, J. Chem. Inf. Model., 2006, ASAP alert. DOI 10.1021/ci600411v
- ^ C. Helma, Predictive Toxicology, CRC Press, 2005.
See also
|





