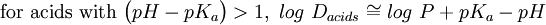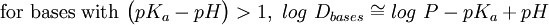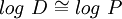To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Partition coefficient
In the fields of organic and medicinal chemistry, a partition (P) or distribution coefficient (D) is the ratio of concentrations of a compound in the two phases of a mixture of two immiscible solvents at equilibrium.[1] Hence these coefficients are a measure of differential solubility of the compound between these two solvents. Normally one of the solvents chosen is water while the second is hydrophobic such as octanol.[2] Hence both the partition and distribution coefficient are measures of how hydrophilic ("water loving") or hydrophobic ("water fearing") a chemical substance is. Partition coefficients are useful for example in estimating distribution of drugs within the body. Hydrophobic drugs with high partition coefficients are preferentially distributed to hydrophobic compartments such as lipid bilayers of cells while hydrophilic drugs (low partition coefficients) preferentially are found in hydrophilic compartments such as blood serum. Product highlight
Partition coefficient and log PThe partition coefficient is the ratio of concentrations of un-ionized compound between the two solutions. To measure the partition coefficient of ionizable solutes, the pH of the aqueous phase is adjusted such that the predominant form of the compound is un-ionized. The logarithm of the ratio of the concentrations of the un-ionized solute in the solvents is called log P:
Distribution coefficient and log DThe distribution coefficient is the ratio of the sum of the concentrations of all forms of the compound (ionized plus unionized) in each of the two phases. For measurements of distribution coefficient, the pH of the aqueous phase is buffered to a specific value such that the pH is not significantly perturbed by the introduction of the compound. The logarithm of the ratio of the sum of concentrations of the solute's various forms in one solvent, to the sum of the concentrations of its forms in the other solvent is called Log D:
In addition, log D is pH dependent, hence the one must specify the pH at which the log D was measured. Of particular interest is the log D at pH = 7.4 (the physiological pH of blood serum). For un-ionizable compounds, log P = log D at any pH. Applications
PharmacologyPharmacokineticsIn the context of pharmacokinetics (what the body does to a drug), the distribution coefficient has a strong influence on ADME properties (Absorption, Distribution, Metabolism, and Excretion) of the drug. Hence the hydrophobicity of a compound (as measured by its distribution coefficient) is a major determinant of how drug-like it is. More specifically, in order for a drug to be orally absorbed, it normally must first pass through lipid bilayers in the intestinal epithelium (a process known as transcellular transport). For efficient transport, the drug must be hydrophobic enough to partition into the lipid bilayer, but not so hydrophobic, that once it is in the bilayer, it will not partition out again.[3] Likewise, hydrophobicity plays a major role in determining where drugs are distributed within the body after adsorption and as a consequence in how rapidly they are metabolized and excreted. PharmacodynamicsIn the context of pharmacodynamics (what a drug does to the body), the hydrophobic effect is the major driving force for the binding of drugs to their receptor targets.[4][5] On the other hand, hydrophobic drugs tend to be more toxic because they in general are retained longer, have a wider distribution within the body (e.g., intracellular), are somewhat less selective in their binding to proteins, and finally are often extensively metabolized. In some cases the metabolites may be chemically reactive. Hence it is advisable to make the drug as hydrophilic as possible while it still retains adequate binding affinity to the therapeutic protein target.[6] Therefore the ideal distribution coefficient for a drug is usually intermediate (not too hydrophobic nor too hydrophilic). Consumer ProductsMany other industries take into account distribution coefficients for example in the formulation of make-up, topical ointments, dyes, hair colors and many other consumer products. AgrochemicalsHydrophobic insecticides and herbicides tend to be more active. On the other hand, hydrophobic agrochemicals in general have longer half lives and therefore display increased risk of adverse environmental impact. EnvironmentalThe hydrophobicity of a compound can give scientists an indication of how easily a compound might be taken up in groundwater to pollute waterways, and its toxicity to animals and aquatic life.[7] Distribution coefficients may be measured or predicted for compounds currently causing problems or with foresight to gauge the structural modifications necessary to make a compound environmentally more friendly in the research phase. In the field of hydrogeology, the octanol water partition coefficient, or Kow, is used to predict and model the migration of dissolved hydrophobic organic compounds in soil and groundwater. MeasurementShake flask (or tube) methodThe classical and most reliable method of log P determination is the shake-flask method, which consists of dissolving some of the solute in question in a volume of octanol and water, then measuring the concentration of the solute in each solvent. The most common method of measuring the distribution of the solute is by UV/VIS spectroscopy. There are a number of pros and cons to this method: Pros:
Cons:
As an alternative to UV/VIS spectroscopy other methods can be used to measure the distribution, one of the best is to use a carrier free radiotracer. In this method (which is well suited for the study of the extraction of metals) a known amount of a radioactive material is added to one of the phases. The two phases are then brought into contact and mixed until equilibrium has been reached. Then the two phases are separated before the radioactivity in each phase is measured. If an energy dispersive detector can be used (such as a high purity germanium detector) then it is possible to use several different radioactive metals at once, with the more simple gamma ray detectors it is only possible to use one radioactive element in the sample. If the volume of both of the phases are the same then the math is very simple. For a hypothetical solute (S) D or P = radioactivity of the organic phase / radioactivity of the aqueous phase D or P = [Sorganic]/[Saqueous] In such an experiment using a carrier free radioisotope the solvent loading is very small, hence the results are different from those which are obtained when the concentration of the solute is very high. A disadvantage of the carrier free radioisotope experiment is that the solute can absorb on the surfaces of the glass (or plastic) equipment or at the interface between the two phases. To guard against this the mass balance should be calculated. It should be the case that radioactivity of the organic phase + radioactivity of the aqueous phase = initial radioactivity of the phase bearing the radiotracer For nonradioactive metals, it is possible in some cases to use ICP-MS or ICP-AES. Sadly ICP methods often suffer from many interferences which do not apply to gamma spectroscopy so hence the use of radio-tracers (counted by gamma ray spectroscopy) is often more straightforward. HPLC determinationA faster method of log P determination makes use of high-performance liquid chromatography. The log P of a solute can be determined by correlating its retention time with similar compounds with known log P values.[8] Pros:
Cons:
Electrochemical methodsIn the recent past some experiments using polarised liquid interfaces have been used to examine the thermodynamics and kinetics of the transfer of charged species from one phase to another. Two main methods exist.
PredictionQSPR (Quantitative Structure-Property Relationship) algorithms calculate a log P in several different ways:
Some Octanol-Water partition coefficient dataThe given values[20] are sorted by the partition coefficient. Acetamide is hydrophilic and 2,2',4,4',5-Pentachlorobiphenyl is lipophilic.
Values for other compounds may be found in Sangster Research Laboratories' database[3]. LimitationsLogP is not an accurate determinant of lipophilicity for ionizable compounds because it only correctly describes the partition coefficient of neutral (uncharged) molecules. Taking the example of drug discovery we see how the limitations of logP can affect research. Since the majority of drugs (approximately 80%) are ionizable, logP is not an appropriate predictor of a compound's behaviour in the changing pH environments of the body. The distribution coefficient (LogD) is the correct descriptor for ionizable systems. See alsoLogP calculatorsThere are many logP calculators or predictors available both commercially and for free.
References
Categories: Medicinal chemistry | Organic chemistry |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Partition_coefficient". A list of authors is available in Wikipedia. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



![log\ P_{oct/wat} = log\Bigg(\frac{\big[solute\big]_{octanol}}{\big[solute\big]_{water}^{un-ionized}}\Bigg)](images/math/8/d/2/8d20065895fcd23c2f7320352432290e.png)
![log\ D_{oct/wat} = log\Bigg(\frac{\big[solute\big]_{octanol}}{\big[solute\big]_{water}^{ionized}+\big[solute\big]_{water}^{un-ionized}}\Bigg)](images/math/f/2/9/f2949789212a9063e235dde8f4ee4ce3.png)
![log\ D_{acids} = log\ P + log\Bigg[\frac{1}{(1+10^{pH-pK_a})}\Bigg]](images/math/1/d/2/1d271a15d9e5bfcea818b489906b44c7.png)
![log\ D_{bases} = log\ P + log\Bigg[\frac{1}{(1+10^{pK_a-pH})}\Bigg]](images/math/7/0/b/70b14db31996c707a40236716853f0c9.png)