To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Car-Parrinello methodThe Car-Parrinello method in computational chemistry is a type of ab initio (first principles) molecular dynamics, usually employing periodic boundary conditions, planewave basis sets, and DFT. In contrast to Born-Oppenheimer molecular dynamics wherein the nuclear (ions) degree of freedom are propagated using ionic forces which are calculated at each iteration by approximately solving the electronic problem with conventional matrix diagonalization methods, the Car-Parrinello method explicitly introduces the electronic degrees of freedom as (fictitious) dynamical variables, writing an extended Lagrangian for the system which leads to a system of coupled equations of motion for both ions and electrons. In this way an explicit electronic minimization at each iteration is not needed: after an initial standard electronic minimization, the fictitious dynamics of the electrons keep them on the electronic ground state corresponding to each new ionic configuration visited along the dynamics, thus yielding accurate ionic forces. In order to maintain this adiabaticity condition, it is necessary that the fictitious mass of the electrons is chosen small enough to avoid a significant energy transfer from the ionic to the electronic degrees of freedom. This small fictitious mass in turn requires that the equations of motion are integrated using a smaller time step than the ones (1-10 fs) commonly used in Born-Oppenheimer molecular dynamics. Product highlight
Fictitious DynamicsLagrangianWhere Orthogonality Constraintwhere δij is the Kronecker Delta function. Equations of MotionYou get the Equations of Motion by taking the stationary point of the Lagrangian with respect to ψi and RI, with the orthogonality constraint. Born-Oppenheimer LimitIn the formal limit where References
See alsoCar-Parrinello Molecular Dynamics, a software package implementing the method. Categories: Molecular dynamics | Quantum chemistry |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Car-Parrinello_method". A list of authors is available in Wikipedia. |



![\mathcal{L} = \frac{1}{2}\left(\sum_I M_I \dot R_I^2 + \mu \int dr |\dot\psi_i|^2 \right) - E\left[\{\psi_i\},\{R_i\}\right]](images/math/d/1/e/d1ec220d4a3b47ede3600ff7efe10b52.png)
![E\left[\{\psi_i\},\{R_i\}\right]](images/math/c/6/8/c68aa8dd772249ff69f62d0ba69ca40d.png) is the Kohn-Sham energy density functional, which outputs energy values when given Kohn-Sham wavefunctions and nuclear positions.
is the Kohn-Sham energy density functional, which outputs energy values when given Kohn-Sham wavefunctions and nuclear positions.
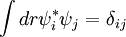
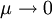 , the Equations of Motion approach Born-Oppenheimer Molecular Dynamics. However numerical implementation limit can lead to inefficient rapid oscillatory trajectories; given the integration framework and according to the problem,
, the Equations of Motion approach Born-Oppenheimer Molecular Dynamics. However numerical implementation limit can lead to inefficient rapid oscillatory trajectories; given the integration framework and according to the problem, Last viewed


