To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Cell listsCell lists (also sometimes referred to as Cell linked-lists) are a tool for finding all atom pairs within a given cut-off distance of each other in Molecular dynamics simulations. These pairs are needed to compute the short-range non-bonded interactions in a system, such as Van der Waals forces or the short-range part of the electrostatic interaction when using Ewald summation.
Product highlight
AlgorithmCell lists work by subdividing the simulation domain into cells with an edge length greater than or equal to the cut-off radius of the interaction to be computed. The particles are sorted into these cells and the interactions are computed between particles in the same or neighbouring cells. In its most basic form, the non-bonded interactions for a cut-off distance rc are computed as follows:
Since the cell length is at least rc in all dimensions, no particles within rc of each other can be missed. Given a simulation with N particles with a homogeneous particle density, the number of cells m is proportional to N and the cut-off radius (i.e. if N increases, so does the number of cells). The average number of particles per cell Periodic Boundary ConditionsIn most simulations, Periodic boundary conditions are used to avoid imposing artificial boundary conditions. Using cell lists, these boundaries can be implemented in two ways Ghost CellsIn the ghost cells approach, the simulation box is wrapped in an additional layer of cells. These cells contain periodically wrapped copies of the corresponding simulation cells inside the domain. Although the data -- and usually also the computational cost -- is doubled for interactions over the periodic boundary, this approach has the advantage of being straight-forward to implement and very easy to parallelize, since cells will only interact with their geographical neighbours. Periodic WrappingInstead of creating ghost cells, cell pairs that interact over a periodic boundary can also use a periodic correction vector
This approach, although more efficient than using ghost cells, is less straight-forward to implement (the cell pairs need to be identified over the periodic boundaries and the vector ImprovementsDespite reducing the computational cost of finding all pairs within a given cut-off distance from Consider a computational cell with edge length equal to the cut-off radius rc. The pairwise distance between all particles in the cell and in one of the neighbouring cells is computed. The cell has 26 neighbours: 6 sharing a common face, 12 sharing a common edge and 8 sharing a common corner. Of all the pairwise distances computed, only about 16% percent will actually less or equal rc. Otherwise put, 84% of all pairwise distance computations are spurious. One way of overcoming this inefficiency is to partition the domain into cells of edge length smaller than rc. The pairwise interactions are then not just computed between neighboring cells, but between all cells within rc of each other (first suggested in [1] and implemented and analysed in [2], [3] and [4]). This approach can be taken to the limit wherein each cell holds at most one single particle, therefore reducing the number of spurious pairwise distance evaluations to zero. This gain in efficiency, however, is quickly offset by the number of cells Cβ that need to be inspected for every interaction with a cell Cα, which grows cubically with the inverse of the cell edge length. Setting the edge length to rc / 2, however, already reduces the number of spurious distance evaluations to 63%. Another approach outlined in [5], where the particles are first sorted along the axis connecting the cell centers. This approach generates only about 40% spurious pairwise distance computations, yet carries an additional cost due to sorting the particles. References
Categories: Molecular dynamics | Computational chemistry | Molecular physics |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Cell_lists". A list of authors is available in Wikipedia. |



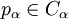 do
do
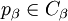 do
do
![r^2 = \| \mathbf x[p_\alpha] - \mathbf x[p_\beta] \|_2^2](images/math/c/a/1/ca14dc40a71d1f3dc52ba5078e682de0.png)
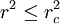 then
then
 therefore does not depend on the total number of particles. The cost of interacting two cells is in
therefore does not depend on the total number of particles. The cost of interacting two cells is in 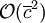 . The number of cell pairs is proportional to the number of cells which is again proportional to the number of particles
. The number of cell pairs is proportional to the number of cells which is again proportional to the number of particles 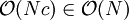 , which is significantly better than computing the
, which is significantly better than computing the 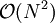 pairwise distances naively.
pairwise distances naively.
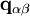 . This vector, which can be stored or computed for every cell pair
. This vector, which can be stored or computed for every cell pair ![r^2 = \| \mathbf x[p_\alpha] - \mathbf x[p_\beta] - \mathbf q_{\alpha\beta} \|^2_2](images/math/b/e/6/be6f673e9c1574fa012cb5c1115f4b0b.png) .
.
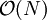 , the cell list algorithm listed above still has some inefficiencies.
, the cell list algorithm listed above still has some inefficiencies.


