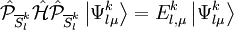To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
N-Electron Valence state Perturbation TheoryIn quantum chemistry, N-Electron Valence state Perturbation Theory (NEVPT) is a perturbative treatment applicable to multireference CASCI-type wavefunctions. It can be considered as a generalization of the well-known second-order Møller-Plesset perturbation theory to multireference Complete Active Space cases. The theory is directly integrated into the quantum chemistry package DALTON. The research performed into the development of this theory lead to various implementations. The theory here presented refers to the deployment for the Single-State NEVPT, where the perturbative correction is applied to a single electronic state. Research implementations has been also developed for Quasi-Degenerate cases, where a set of electronic states undergo the perturbative correction at the same time, allowing interaction among themselves. The theory development makes use of the quasi-degenerate formalism by Lindgren and the Hamiltonian multipartitioning technique from Zaitsevskii and Malrieu. Product highlight
TheoryLet obtained diagonalizing the true Hamiltonian where then the perturber wavefunctions can be written as The pattern of inactive orbitals involved in the procedure can be grouped as a collective index l, so to represent the various perturber wavefunctions as Supposing indexes i and j referring to core orbitals, a and b referring to active orbitals and r and s referring to virtual orbitals, the possible excitation schemes are:
These cases always represent situations where interclass electronic excitations happen. Other three excitation schemes involve a single interclass excitation plus an intraclass excitation internal to the active space:
Totally uncontracted approachA possible approach is to define the perturber wavefunctions into Hilbert spaces The full dimensionality of these spaces can be exploited to obtain the definition of the perturbers, by diagonalizing the Hamiltonian inside them This procedure is impractical given its high computational cost: for each stripping out the constant contribution of the inactive part and leaving a subsystem to be solved for the valence part The total energy Strongly Contracted ApproachA different choice in the development of the NEVPT approach is to choose a single function for each space where For each space, appropriate operators can be devised. We will not present their definition, as it could result overkilling. Suffice to say that the resulting perturbers are not normalized, and their norm plays an important role in the Strongly Contracted development. To evaluate these norms, the spinless density matrix of rank not higher than three between the An important property of the where The expression for the first-order correction to the wavefunction is therefore and for the energy is It is important to note that this result still misses a definition of the perturber energies leading to Developing the first term and extracting the inactive part of the Dyall's Hamiltonian it can be obtained with Δεl equal to the sum of the orbital energies of the newly occupied virtual orbitals minus the orbital energies of the unoccupied core orbitals. The term that still need to be evaluated is the braket involving the commutator. This can be obtained developing each V operator and substituting. To obtain the final result is necessary to evaluate Koopmans matrices and density matrices involving only active indexes. An interesting case is represented by the contribution for the NEVPT2 can therefore be seen as a generalized form of MP2 to multireference wavefunctions. Partially Contracted ApproachAn alternative approach, named Partially Contracted (PC) is to define the perturber wavefunctions in a subspace The Partially Contracted approach makes use of functions Once all the As usual, the evaluation of the Partially Contracted perturbative correction by means of the Dyall Hamiltonian involves simply manageable entities for nowadays computers. Although the Strongly Contracted approach makes use of a perturbative space with very low flexibility, in general it provides values in very good agreement with those obtained by the more decontracted space defined for the Partially Contracted approach. This can be probably explained by the fact that the Strongly Contracted perturbers are a good average of the totally decontracted perturbative space. It should also be noted that the Partially Contracted evaluation has a very little overhead in computational cost with respect to the Strongly Contracted one, therefore they are normally evaluated together. PropertiesNEVPT is blessed with many important properties, making the approach very solid and reliable. These properties arise both from the theoretical approach used and on the Dyall's Hamiltonian particular structure:
See also
References
|
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "N-Electron_Valence_state_Perturbation_Theory". A list of authors is available in Wikipedia. |



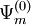 be a zero-order CASCI wavefunction, defined as a linear combination of Slater determinants
be a zero-order CASCI wavefunction, defined as a linear combination of Slater determinants
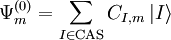
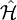 inside the CASCI space
inside the CASCI space
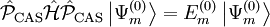
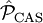 is the projector inside the CASCI space.
It is possible to define perturber wavefunctions in NEVPT as zero-order wavefunctions of the outer space (external to CAS) where
is the projector inside the CASCI space.
It is possible to define perturber wavefunctions in NEVPT as zero-order wavefunctions of the outer space (external to CAS) where 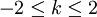 . Decomposing the zero-order CASCI wavefunction as an antisymmetrized product of the inactive part
. Decomposing the zero-order CASCI wavefunction as an antisymmetrized product of the inactive part 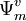
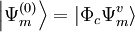
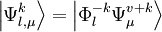
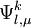 , with
, with 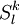 defined by those determinants with given k and l labels. It is interesting to note that the determinants characterizing these spaces can be written as a partition comprising the same inactive (core + virtual) part
defined by those determinants with given k and l labels. It is interesting to note that the determinants characterizing these spaces can be written as a partition comprising the same inactive (core + virtual) part 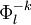 and all possible valence (active) parts
and all possible valence (active) parts 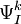
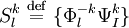
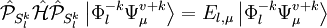
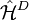 . This Hamiltonian behaves like the true Hamiltonian inside the CAS space, having the same eigenvalues and eigenvectors of the true Hamiltonian projected onto the CAS space. Also, given the decomposition for the wavefunction defined before, the action of the Dyall's Hamiltonian can be partitioned into
. This Hamiltonian behaves like the true Hamiltonian inside the CAS space, having the same eigenvalues and eigenvectors of the true Hamiltonian projected onto the CAS space. Also, given the decomposition for the wavefunction defined before, the action of the Dyall's Hamiltonian can be partitioned into
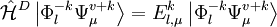
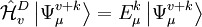
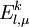 is the sum of
is the sum of 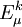 and the energies of the orbitals involved in the definition of the inactive part
and the energies of the orbitals involved in the definition of the inactive part 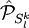 of the application of the Hamiltonian to the contracted zero order wavefunction. In other words
of the application of the Hamiltonian to the contracted zero order wavefunction. In other words
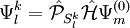
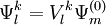
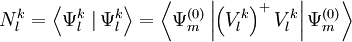
 is that any other function of the space
is that any other function of the space 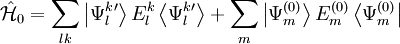
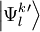 are the normalized
are the normalized 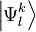 .
.
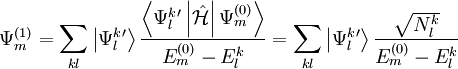
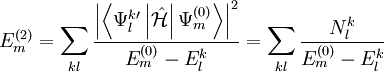
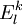 , which can be defined in a computationally advantageous approach by means of the Dyall's Hamiltonian
, which can be defined in a computationally advantageous approach by means of the Dyall's Hamiltonian
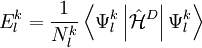
![N_{l}^{k} E_{l}^{k} = \left\langle \Psi_{m}^{(0)} \left| \left( V_{l}^{k}\right)^{+} \hat{\mathcal{H}}^D V_{l}^{k} \right| \Psi_{m}^{(0)} \right\rangle = \left\langle \Psi_{m}^{(0)} \left| \left(V_{l}^{k} \right)^{+} V_{l}^{k} \hat{\mathcal{H}}^D \right| \Psi_{m}^{(0)} \right\rangle + \left\langle \Psi_{m}^{(0)} \left| \left( V_{l}^{k}\right)^{+} \left[ \hat{\mathcal{H}}^D , V_{l}^{k} \right] \right| \Psi_{m}^{(0)} \right\rangle](images/math/0/6/4/0640c345b21f0d2959bb62656e259672.png)
![E_{l}^{k} = E_m^{(0)} + \Delta \epsilon_l + \frac{1}{N_l^k} \left\langle \Psi_{m}^{(0)} \left| \left(V_{l}^{k}\right)^{+} \left[\hat{\mathcal{H}}_v , V_{l}^{k} \right] \right| \Psi_{m}^{(0)} \right\rangle](images/math/b/d/b/bdb2caf57cf6e98910511fa0e7db7eae.png)
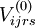 case, which is trivial and can be demonstrated identical to the Møller-Plesset second-order contribution
case, which is trivial and can be demonstrated identical to the Møller-Plesset second-order contribution
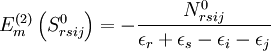
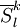 of
of 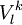 operators, after decontraction of their formulation. For example, in the case of the
operators, after decontraction of their formulation. For example, in the case of the 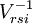 operator
operator
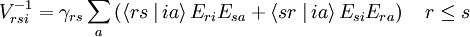
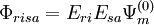 and
and 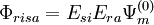 . These functions must be orthonormalized and purged of linear dependencies which may arise. The resulting set spans the
. These functions must be orthonormalized and purged of linear dependencies which may arise. The resulting set spans the 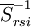 space.
space.