To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
OPLSThe OPLS (Optimized Potential for Liquid Simulations) force field was developed by Prof. William L. Jorgensen at Purdue University and later at Yale University. Product highlight
Functional formThe functional form of the OPLS force field is very similar to that of AMBER.
Intramolecular non-bonded interactions (Eab) are only counted for atoms three or more bonds apart; 1, 4 interactions are scaled down by fij = 0.5; otherwise, fij = 1.0. All the interaction sites are centered on the atoms; there are no "lone pairs". ParameterizationSeveral sets of OPLS parameters have been published. There is OPLS-ua (united atom), which includes hydrogen atoms next to carbon implicitly in the carbon parameters, and can be used to save simulation time. OPLS-aa (all atom) includes every atom explicitly. Later publications include parameters for other specific functional groups and types of molecules such as carbohydrates. OPLS simulations in aqueous solution typically use the TIP4P or TIP3P water model. A distinctive feature of the OPLS parameters is that they were optimized to fit experimental properties of liquids, such as density and heat of vaporization, in addition to fitting gas-phase torsional profiles. ImplementationThe reference implementations of the OPLS force field are the BOSS and MCPRO programs developed by Jorgensen. Other packages such as TINKER and GROMACS also implement OPLS force fields. References
|
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "OPLS". A list of authors is available in Wikipedia. |



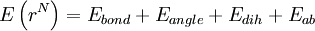
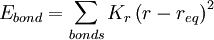
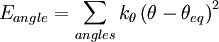
![E_{dih} = \frac {V_1} {2} \left [ 1 + \cos \left ( \phi \right ) \right ] + \frac {V_2} {2} \left [ 1 - \cos \left ( 2 \phi \right ) \right ] + \frac {V_3} {2} \left [ 1 + \cos \left ( 3 \phi \right ) \right ] + \frac {V_4} {2} \left [ 1 - \cos \left ( 4 \phi \right ) \right ]](images/math/8/a/4/8a44b9aafd75d9b15c6aeb89c8298445.png)
![E_{ab} = \sum_{i} ^{on\ a} \sum_{j} ^{on\ b} \left \{ 4 \epsilon_{ij} \left [ \left( \frac {\sigma_{ij}}{r_{ij}} \right )^{12} - \left ( \frac {\sigma_{ij}}{r_{ij}} \right )^6 \right ] + \frac {q_iq_j e^2}{r_{ij}} \right \} f_{ij}](images/math/2/8/1/281becd19b86edc12f7e4dbc795c6d76.png)
Last viewed


