To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
AMBER
AMBER (an acronym for Assisted Model Building and Energy Refinement) is a family of force fields for molecular dynamics of biomolecules originally developed by the late Peter Kollman's group at the University of California, San Francisco. AMBER is also the name for the molecular dynamics simulation package that implements these force fields. It is maintained by an active collaboration between David Case at The Scripps Research Institute, Tom Cheatham at the University of Utah, Tom Darden at NIEHS, Ken Merz at Florida, Carlos Simmerling at Stony Brook University, Ray Luo at UC Irvine, and Junmei Wang at Encysive Pharmaceuticals. Product highlight
Force FieldThe term "AMBER force field" generally refers to the functional form used by the family of AMBER force fields. This form includes a number of parameters; each member of the family of AMBER force fields provides values for these parameters and has its own name. Functional formThe functional form of the AMBER force field is Note that despite the term force field, this equation defines the potential energy of the system; the force is the derivative of this potential with respect to position. The meanings of right hand side terms are:
Parameter setsTo use the AMBER force field, it is necessary to have values for the parameters of the force field (e.g. force constants, equilibrium bond lengths and angles, charges). A fairly large number of these parameter sets exist, and are described in detail in the AMBER software user manual. Each parameter set has a name, and provides parameters for certain types of molecules.
SoftwareThe AMBER software suite provides a set of programs for applying the AMBER forcefields to simulations of biomolecules. It is written in Fortran 90 and C with support for most major Unix-like systems and compilers. Development is conducted by a loose association of mostly academic labs. New versions are generally released in March of even numbered years; AMBER 9 was released in March 2006. The software is available under a site-license agreement, which includes full source, currently priced at US$400 for non-commercial and US$20,000 for commercial organizations. Programs
See alsoReferences
Categories: Molecular dynamics software | Force fields |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "AMBER". A list of authors is available in Wikipedia. |



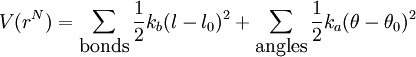
![+ \sum_\mbox{torsions} \frac{1}{2} V_n [1+\cos(n \omega- \gamma)]](images/math/1/5/0/15016a1049863d779ef77d4b73217f06.png)
![+\sum_{j=1} ^{N-1} \sum_{i=j+1} ^N \left\{4\epsilon_{i,j}\left[\left(\frac{\sigma_{ij}}{r_{ij}} \right)^{12} - \left(\frac{\sigma_{ij}}{r_{ij}} \right)^6 \right]+ \frac{q_iq_j}{4\pi \epsilon_0 r_{ij}}\right\}](images/math/8/e/d/8ed9a67e2b171fc91cd5750fa41cb973.png)


