To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Force field (chemistry)
In the context of molecular mechanics, a force field (also called a forcefield) refers to the functional form and parameter sets used to describe the potential energy of a system of particles (typically but not necessarily atoms). Force field functions and parameter sets are derived from both experimental work and high-level quantum mechanical calculations. "All-atom" force fields provide parameters for every atom in a system, including hydrogen, while "united-atom" force fields treat the hydrogen and carbon atoms in methyl and methylene groups as a single interaction center. "Coarse-grained" force fields, which are frequently used in long-time simulations of proteins, provide even more abstracted representations for increased computational efficiency. The usage of the term "force field" in chemistry and computational biology differs from the standard usage in physics. In chemistry usage a force field is defined as a potential function, while the term is used in physics to denote the negative gradient of a scalar potential. Product highlightFunctional form
The basic functional form of a force field encapsulates both bonded terms relating to atoms that are linked by covalent bonds, and nonbonded (also called "noncovalent") terms describing the long-range electrostatic and van der Waals forces. The specific decomposition of the terms depends on the force field, but a general form for the total energy can be written as
The bond and angle terms are usually modeled as harmonic oscillators in force fields that do not allow bond breaking. The functional form is highly variable. It can include potentials for hydrogen bonds, an "improper torsion" term to account for the planarity of aromatic rings and other conjugated systems, and "cross-terms" that describe coupling of different internal variables, such as dihedral angles and bond lengths. The nonbonded terms are most computationally intensive because they include many more pairwise interactions per atom. The van der Waals term is usually computed with a Lennard-Jones potential and the electrostatic term with Coulomb's law, although both can be buffered or scaled by a constant factor to produce better agreement with experimental observation. ParameterizationIn addition to the functional form of the potentials, a force field defines a set of parameters for each type of atom. For example, a force field would include distinct parameters for an oxygen atom in a carbonyl functional group and in a hydroxyl group. The typical parameter set includes values for atomic mass, van der Waals radius, and partial charge for individual atoms, and equilibrium values of bond lengths, bond angles, and dihedral angles for pairs, triplets, and quandruplets of bonded atoms, and values corresponding to the effective spring constant for each potential. Most current force fields use a "fixed-charge" model by which each atom is assigned a single value for the atomic charge that is not affected by the local electrostatic environment; proposed developments in next-generation force fields incorporate models for polarizability, in which a particle's charge is influenced by electrostatic interactions with its neighbors. For example, polarizability can be approximated by the introduction of induced dipoles; it can also be represented by Drude particles, or massless, charge-carrying virtual sites attached by a springlike harmonic potential to each polarizable atom. The introduction of polarizability into force fields in common use has been inhibited by the high computational expense associated with calculating the local electrostatic field. Although many molecular simulations involve biological macromolecules such as proteins, DNA, and RNA, the parameters for given atom types are generally derived from observations on small organic molecules that are more tractable for experimental studies and quantum calculations. Different force fields can be derived from dissimilar types of experimental data, such as enthalpy of vaporization (OPLS), enthalpy of sublimation (CFF), dipole moments, or various spectroscopic parameters (CFF). Parameter sets and functional forms are defined by force field developers to be self-consistent. Because the functional forms of the potential terms vary extensively between even closely related force fields (or successive versions of the same force field), the parameters from one force field should never be used in conjunction with the potential from another. DeficienciesAll force fields are based on numerous approximations and derived from different types of experimental data. Therefore they are called empirical. Some existing force fields usually do not account for electronic polarization of the environment, an effect that can significantly reduce electrostatic interactions of partial atomic charges. This problem was addressed by developing “polarizable force fields” [1] [2] or using macroscopic dielectric constant. However, application of a single value of dielectric constant is questionable in the highly heterogeneous environments of proteins or biological membranes, and the nature of the dielectric depends on the model used [3]. All types of van der Waals forces are also strongly environment-dependent, because these forces originate from interactions of induced and “instantaneous” dipoles (see Intermolecular force). The original Fritz London theory of these forces can only be applied in vacuum. A more general theory of van der Waals forces in condensed media was developed by A. D. McLachlan in 1963 (this theory includes the original London’s approach as a special case) [4]. The McLachlan theory predicts that van der Waals attractions in media are weaker than in vacuum and follow the "like dissolves like" rule, which means that different types of atoms interact more weakly than identical types of atoms. [5]. This is in contrast to “combinatorial rules” or Slater-Kirkwood equation applied for development of the classical force fields. The “combinatorial rules” state that interaction energy of two dissimilar atoms (e.g. C…N) is an average of the interaction energies of corresponding identical atom pairs (i.e. C…C and N…N). According to McLachlan theory, the interactions of particles in a media can even be completely repulsive, as observed for liquid helium [4]. The conclusions of McLachlan theory are supported by direct measurements of attraction forces between different materials (Hamaker constant), as explained by Jacob Israelachvili in his book "Intermolecular and surface forces". It was concluded that "the interaction between hydrocarbons across water is about 10% of that across vacuum" [4]. Such effects are unaccounted in the standard molecular mechanics. Another round of criticism came from practical applications, such as protein structure refinement. It was noted that CASP participants did not try to refine their models to avoid "a central embarrassment of molecular mechanics, namely that energy minimization or molecular dynamics generally leads to a model that is less like the experimental structure". [6] Actually, the force fields have been successfully applied for protein structure refinement in different X-ray crystallography and NMR spectroscopy applications, especially using program XPLOR [7]. However, such refinement is driven primarily by a set of experimental constraints, whereas the force fields serve merely to remove interatomic hindrances. The results of calculations are practically the same with rigid sphere potentials implemented in program DYANA [8] (calculations from NMR data), or with programs for crystallographic refinement that do not use any energy functions. The deficiencies of the force fields remain a major bottleneck in homology modeling of proteins [9]. Such situation gave rise to development of alternative empirical scoring functions specifically for ligand docking [10], protein folding [11] [12] [13], computational protein design [14] [15] [16], and modeling of proteins in membranes [17]. There is also an opinion that molecular mechanics may operate with energy which is irrelevant to protein folding or ligand binding [18]. The parameters of typical force fields reproduce enthalpy of sublimation, i.e. energy of evaporation of molecular crystals. However, it was recognized that protein folding and ligand binding are thermodynamically very similar to crystallization, or liquid-solid transitions, because all these processes represent “freezing” of mobile molecules in condensed media [19] [20] [21]. Therefore, free energy changes during protein folding or ligand binding are expected to represent a combination of an energy similar to heat of fusion (energy absorbed during melting of molecular crystals), a conformational entropy contribution, and solvation free energy. The heat of fusion is significantly smaller than enthalpy of sublimation [4]. Hence, the potentials describing protein folding or ligand binding must be weaker than potentials in molecular mechanics. Indeed, the energies of H-bonds in proteins are ~ -1.5 kcal/mol when estimated from protein engineering or alpha helix to coil transition data [22] [23], but the same energies estimated from sublimation enthalpy of molecular crystals were -4 to -6 kcal/mol [24]. The depths of modified Lennard-Jones potentials derived from protein engineering data were also smaller than in typical force fields and followed the “like dissolves like” rule, as predicted by McLachlan theory [18]. Popular force fieldsDifferent force fields are designed for different purposes. MM2 was developed primarily for conformational analysis of small organic molecules. It is designed to reproduce the equilibrium covalent geometry of molecules as precisely as possible. It implements a large set of parameters that is continuously refined and updated for many different classes of organic compounds (MM3 and MM4). CFF was developed by Warshel, Lifson and coworkers as a general method for unifying studies of energies, structures and vibration of general molecules and molecular crystals. The CFF program, developed by Levitt and Warshel, is based on the Cartesian representation of all the atoms, and it served as the basis for many subsequent simulation programs. ECEPP was developed specifically for modeling of peptides and proteins. It uses fixed geometries of amino acid residues to simplify the potential energy surface. Thus, the energy minimization is conducted in the space of protein torsion angles. Both MM2 and ECEPP include potentials for H-bonds and torsion potentials for describing rotations around single bonds. ECEPP/3 was implemented (with some modifications) in Internal Coordinate Mechanics and FANTOM [25]. AMBER, CHARMM and GROMOS have been developed primarily for molecular dynamics of macromolecules, although they are also commonly applied for energy minimization. Therefore, the coordinates of all atoms are considered as free variables. Classical force fields
Second-generation force fields
Polarizable force field based on induced dipole
Polarizable Force Fields based on point charges
Polarizable Force Fields based on distributed multipoles
Polarizable Force Fields based on density
Reactive Force Fields
Other
Water ModelsMain article: water model The set of parameters used to model water or aqueous solutions (basically a force field for water) is called a water model. Water has attracted a great deal of attention due to its unusual properties and its importance as a solvent. Many water models have been proposed; some examples are TIP3P, TIP4P, SPC, and ST2. See also
References
Further reading
Categories: Force fields | Intermolecular forces | Molecular physics |
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Force_field_(chemistry)". A list of authors is available in Wikipedia. |



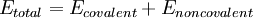 where the components of the covalent and noncolvalent contributions are given by the following summations:
where the components of the covalent and noncolvalent contributions are given by the following summations: