To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Alpha helix
A common motif in the secondary structure of proteins, the alpha helix (α-helix) is a right-handed coiled conformation, resembling a spring, in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier ( Product highlight
Historical developmentIn the early 1930s, William Astbury showed that there were drastic changes in the X-ray fiber diffraction of moist wool or hair fibers upon significant stretching. The data suggested that the unstretched fibers had a coiled molecular structure with a characteristic repeat of ~5.1 Å (= 0.51 nm). Astbury proposed that (1) the unstretched protein molecules formed a helix (which he called the α-form); and (2) the stretching caused the helix to uncoil, forming an extended state (which he called the β-form). Although incorrect in their details, Astbury's models of these forms were correct in essence and correspond to modern elements of secondary structure, the α-helix and the β-strand (Astbury's nomenclature was kept), which were developed by Linus Pauling, Robert Corey and Herman Branson in 1951 (see below). Hans Neurath was the first to show that Astbury's models could not be correct in detail, because they involved clashes of atoms.[1] Interestingly, Neurath's paper and Astbury's data inspired H. S. Taylor,[2] Maurice Huggins[3] and Bragg and collaborators[4] to propose models of keratin that resemble the modern α-helix. Two key developments in the modeling of the modern α-helix were (1) the correct bond geometry, thanks to the crystal structure determinations of amino acids and peptides and Pauling's prediction of planar peptide bonds; and (2) the relinquishing of the assumption of an integral number of residues per turn of the helix. The pivotal moment came in January 1948, when Pauling caught a cold and went to bed. Being bored, he drew a polypeptide chain of roughly correct dimensions on a strip of paper and folded it into a helix, being careful to maintain the planar peptide bonds. After a few attempts, he produced a model with physically plausible hydrogen bonds. Pauling then worked with Corey and Branson to confirm his model before publication.[5] StructureGeometry and hydrogen bonding
The amino acids in an α helix are arranged in a right-handed helical structure, 5.4 Å (= 0.54 nm) wide. Each amino acid corresponds to a 100° turn in the helix (i.e., the helix has 3.6 residues per turn), and a translation of 1.5 Å (= 0.15 nm) along the helical axis. Most importantly, the N-H group of an amino acid forms a hydrogen bond with the C=O group of the amino acid four residues earlier; this repeated Residues in α-helices typically adopt backbone (φ, ψ) dihedral angles around (-60°, -45°). More generally, they adopt dihedral angles such that the ψ dihedral angle of one residue and the φ dihedral angle of the next residue sum to roughly -105°. Consequently, α-helical dihedral angles generally fall on a diagonal stripe on the Ramachandran plot (of slope -1), ranging from (-90°, -15°) to (-35°, -70°). For comparison, the sum of the dihedral angles for a 310 helix is roughly -75°, whereas that for the π-helix is roughly -130°. The general formula for the rotation angle Ω per residue of any polypeptide helix with trans isomers is given by the equation[citation needed] The α-helix is tightly packed; there is almost no free space within the helix. The amino-acid side chains are on the outside of the helix, and point roughly "downwards" (i.e., towards the N-terminus), like the branches of an evergreen tree (Christmas tree effect). This directionality is sometimes used in preliminary, low-resolution electron-density maps to determine the direction of the protein backbone. StabilityHelices observed in proteins can range from four to over forty residues long, but a typical helix contains about ten amino acids (about three turns). Short polypeptides generally do not exhibit much alpha helical structure in solution, since the entropic cost associated with the folding of the polypeptide chain is not compensated by sufficiently many stabilizing interactions. The backbone hydrogen bonds of α-helices are generally considered slightly weaker than those found in β-sheets, and are readily attacked by the ambient water molecules. However, in more hydrophobic environments such as the plasma membrane, or in the presence of co-solvents such as trifluoroethanol (TFE), oligopeptides readily adopt stable α-helical structure. Experimental determinationSince the α-helix is defined by its hydrogen bonds, the most reliable experimental methods for determining an α-helix involve an atomic-resolution structure provided by X-ray crystallography or NMR spectroscopy. In some cases, the individual hydrogen bonds can be observed directly as a small scalar coupling in NMR. There are several lower-resolution methods for assigning general helical structure. The NMR chemical shifts (particularly of the Cα, Cβ and C' atoms) and residual dipolar couplings are often characteristic of helices. The far-UV (170-250 nm) circular dichroism spectrum of helices is also idiosyncratic, exhibiting a pronounced double minimum at ~208 nm and ~222 nm. Infrared spectroscopy is rarely used, since the α-helical spectrum resembles that for random coil (although these might be discerned by, e.g., hydrogen-deuterium exchange). Finally, cryo electron microscopy is now capable of discerning individual α-helices within a protein, although their assignment to residues is still an active area of research. Long homopolymers of amino acids often form helices (if soluble). Such long, isolated helices can also be detected by other methods, such as dielectric relaxation, flow birefringence and measurements of its diffusion constant. Strictly speaking, these methods only detect the characteristic prolate (long cigar-like) hydrodynamic shape of a helix, or its large dipole moment. Amino-acid propensitiesDifferent amino-acid sequences have different propensities for forming α-helical structure. Methionine, alanine, leucine, glutamate, and lysine ("MALEK" in the amino-acid 1-letter codes) all have especially high helix-forming propensities, whereas proline, glycine, tyrosine and serine have poor helix-forming propensities. Proline tends to break or kink helices because it cannot donate an amide hydrogen bond (having no amide hydrogen), and because its sidechain interferes sterically; its ring structure also restricts its backbone φ dihedral angle to the vicinity of -70°, which is less common in α-helices. However, proline is often seen as the first residue of a helix, presumably due to its structural rigidity. At the other extreme, glycine also tends to disrupt helices because its high conformational flexibility makes it entropically expensive to adopt the relatively constrained α-helical structure. Dipole momentA helix has an overall dipole moment caused by the aggregate effect of all the individual dipoles from the carbonyl groups of the peptide bond pointing along the helix axis. This can lead to destabilization of the helix through entropic effects. As a result, α helices are often capped at the N-terminal end by a negatively charged amino acid, such as glutamic acid, in order to neutralize this helix dipole. Less common (and less effective) is C-terminal capping with a positively charged amino acid, such as lysine. The N-terminal positive charge is commonly used to bind negatively charged ligands such as phosphate groups, which is especially effective because the backbone amides can serve as hydrogen bond donors. Larger-scale assembliesMyoglobin, the first protein whose structure was solved by X-ray crystallography, is made up of about 70% α helix, with the rest being loops or disordered regions. In classifying proteins by their dominant fold, the Structural Classification of Proteins database maintains a category specifically for all-α proteins. Coiled-coil α helices are highly stable forms in which two or more helices wrap around each other in a "supercoil" structure. Coiled coils contain a highly characteristic sequence motif known as a heptad repeat, in which the motif repeats itself every seven residues along the sequence. The first and especially the fourth residues (known as the a and h positions) are almost always hydrophobic (the fourth residue is typically leucine) and pack together in the interior of the helix bundle. The fifth and seventh residues (the e and g positions) generally have opposing charges and form a salt bridge stabilized by electrostatic interactions. Fibrous proteins such as keratin and myosin often adopt coiled-coil structures, as do several dimerizing proteins. A pair of coiled-coils - a four-helix bundle - is a very common structural motif in proteins. For example, it occurs in human growth hormone and several varieties of cytochrome. The Rop protein, which promotes plasmid replication in bacteria, is an interesting case in which a single polypeptide forms a coiled-coil and two monomers assemble to form a four-helix bundle. The amino acids that make up a particular helix can be plotted on a helical wheel, a representation that illustrates the orientations of the constituent amino acids. Often in globular proteins, as well as in specialized structures such as coiled-coils and leucine zippers, an alpha helix will exhibit two "faces" - one containing predominantly hydrophobic amino acids oriented toward the interior of the protein, in the hydrophobic core, and one containing predominantly polar amino acids oriented toward the solvent-exposed surface of the protein. Functional rolesα helices have particular significance in DNA binding motifs, including helix-turn-helix motifs, leucine zipper motifs and zinc finger motifs. This is because of the convenient structural fact that the diameter of the α helix is 1.2 nanometres, the same as the width of the major groove in B-form DNA. Helix-coil transitionHomopolymers of amino-acids (such as poly-lysine) can adopt α-helical structure at low temperature that is "melted out" at high temperatures. This helix-coil transition was once thought to be analogous to protein denaturation. The statistical mechanics of this transition can be modeled using an elegant transfer matrix method, characterized by two parameters: the propensity to initiate a helix and the propensity to extend a helix. The α-helix in fine artAt least two artists have made explicit reference to the α-helix in their work, Julie Newdoll in painting and Julian Voss-Andreae in sculpture. Bay-Area artist Julie Newdoll, who holds a degree in Microbiology, and a minor in art, has specialized in paintings inspired by microscopic images and molecules since 1990. Her painting "Rise of the Alpha Helix" (2003) features human figures arranged in an α helical arrangement. According to the artist, "the flowers reflect the various types of sidechains that each amino acid holds out to the world".
Julian Voss-Andreae is a German-born sculptor with degrees in experimental physics and sculpture. Since 2001 Voss-Andreae creates "protein sculptures"[6] based on protein structure with the α-helix being one of his preferred objects. Voss-Andreae has made α-helix sculptures from diverse materials including bamboo and whole trees. A monument Voss-Andreae created in 2004 to celebrate the memory of Linus Pauling, the discoverer of the α-helix, is fashioned from a large steel beam rearranged in the structure of the α-helix. The 10' (3 m) tall, bright-red sculpture stands in front of Pauling's childhood home in Portland, Oregon. See also
References and footnotes
Additional references
|
|||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Alpha_helix". A list of authors is available in Wikipedia. | |||||||||||||||||||||||||||||
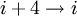 hydrogen bonding). (See also helix.)
hydrogen bonding). (See also helix.)



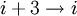 hydrogen bonding) and the π-helix (
hydrogen bonding) and the π-helix (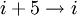 hydrogen bonding). These alternative helices are relatively rare, although the 310 helix is often found at the ends of α-helices, "closing" them off. Transient
hydrogen bonding). These alternative helices are relatively rare, although the 310 helix is often found at the ends of α-helices, "closing" them off. Transient 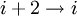 helices (sometimes called δ-helices) have also been reported as intermediates in
helices (sometimes called δ-helices) have also been reported as intermediates in ![3 \cos \Omega = 1 - 4 \cos^{2} \left[\left(\phi + \psi \right)/2 \right]](images/math/8/f/5/8f504de02eb71fe4fabe5d44c83da475.png)
Last viewed


