To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Secondary structureIn biochemistry and structural biology, secondary structure is the general three-dimensional form of local segments of biopolymers such as proteins and nucleic acids (DNA/RNA). It does not, however, describe specific atomic positions in three-dimensional space, which are considered to be tertiary structure. Secondary structure is formally defined by the hydrogen bonds of the biopolymer, as observed in an atomic-resolution structure. In proteins, the secondary structure is defined by patterns of hydrogen bonds between backbone amide groups (sidechain-mainchain and sidechain-sidechain hydrogen bonds are irrelevant), where the DSSP definition of a hydrogen bond is used. In nucleic acids, the secondary structure is defined by the hydrogen bonding between the nitrogenous bases. The hydrogen bonding is correlated with other structural features, however, which has given rise to less formal definitions of secondary structure. For example, residues in protein helices generally adopt backbone dihedral angles in a particular region of the Ramachandran plot; thus, a segment of residues with such dihedral angles is often called a "helix", regardless of whether it has the correct hydrogen bonds. Many other less formal definitions have been proposed, often applying concepts from the differential geometry of curves, such as curvature and torsion. Least formally, structural biologists solving a new atomic-resolution structure will sometimes assign its secondary structure "by eye" and record their assignments in the corresponding PDB file. The rough secondary-structure content of a biopolymer (e.g., "this protein is 40% α-helix and 20% β-sheet.") can often be estimated spectroscopically. For proteins, a common method is far-ultraviolet (far-UV, 170-250 nm) circular dichroism. A pronounced double minimum at 208 and 222 nm indicate α-helical structure, whereas a single minimum at 204 nm or 217 nm reflects random-coil or β-sheet structure, respectively. A less common method is infrared spectroscopy, which detects differences in the bond oscillations of amide groups due to hydrogen-bonding. Finally, secondary-structure contents may be estimated accurately using the chemical shifts of an unassigned NMR spectrum. Secondary structure was introduced by Kaj Ulrik Linderstrøm-Lang in the 1952 Lane medical lectures at Stanford. Product highlight
ProteinsSecondary structure in proteins consists of local inter-residue interactions mediated by hydrogen bonds. The most common secondary structures are alpha helices and beta sheets. Other helices, such as the 310 helix and π helix, are calculated to have energetically favorable hydrogen-bonding patterns but are rarely if ever observed in natural proteins except at the ends of α helices due to unfavorable backbone packing in the center of the helix. Other extended structures such as the polyproline helix and alpha sheet are rare in native state proteins but are often hypothesized as important protein folding intermediates. Tight turns and loose, flexible loops link the more "regular" secondary structure elements. The random coil is not a true secondary structure, but is the class of conformations that indicate an absence of regular secondary structure. Amino acids vary in their ability to form the various secondary structure elements. Proline and glycine are sometimes known as "helix breakers" because they disrupt the regularity of the α helical backbone conformation; however, both have unusual conformational abilities and are commonly found in turns. Amino acids that prefer to adopt helical conformations in proteins include methionine, alanine, leucine, glutamate and lysine ("MALEK" in amino-acid 1-letter codes); by contrast, the large aromatic residues (tryptophan, tyrosine and phenylalanine) and Cβ-branched amino acids (isoleucine, valine, and threonine) prefer to adopt β-strand conformations. However, these preferences are not strong enough to produce a reliable method of predicting secondary structure from sequence alone. The DSSP code
The DSSP code is frequently used to describe the protein secondary structures with a single letter code. DSSP is an acronym for "Dictionary of Protein Secondary Structure", which was the title of the original article actually listing the secondary structure of the proteins with known 3D structure (Kabsch and Sander 1983). The secondary structure is assigned based on hydrogen bonding patterns as those initially proposed by Pauling et al. in 1951 (before any protein structure had ever been experimentally determined).
In DSSP, residues which are not in any of the above conformations is designated as ' ' (space), which sometimes gets designated with C (coil) or L (loop). The helices (G,H and I) and sheet conformations are all required to have a reasonable length. This means that 2 adjacent residues in the primary structure must form the same hydrogen bonding pattern. If the helix or sheet hydrogen bonding pattern is too short they are designated as T or B, respectively. Other protein secondary structure assignment categories exist (sharp turns, Omega loops etc.), but they are less frequently used. DSSP H-bond definitionSecondary structure is defined by hydrogen bonding, so the exact definition of a hydrogen bond is critical. The standard H-bond definition for secondary structure is that of DSSP, which is a purely electrostatic model. It assigns charges of According to DSSP, an H-bond exists if and only if E is less than -0.5 kcal/mol. Although the DSSP formula is a relatively crude approximation of the physical H-bond energy, it is generally accepted as a tool for defining secondary structure. Protein secondary-structure predictionEarly methods of secondary-structure prediction were based on the helix- or sheet-forming propensities of individual amino acids, sometimes coupled with rules for estimating the free energy of forming secondary structure elements. Such methods are typically ~60% accurate in predicting which of the three states (helix/sheet/coil) a residue adopts. A significant increase in accuracy (to nearly ~80%) was made by exploiting multiple sequence alignment; knowing the full distribution of amino acids that occur at a position (and in its vicinity, typically ~7 residues on either side) throughout evolution provides a much better picture of the structural tendencies near that position. For illustration, a given protein might have a glycine at a given position, which by itself might suggest a random coil there. However, multiple sequence alignment might reveal that helix-favoring amino acids occur at that position (and nearby positions) in 95% of homologous proteins spanning nearly a billion years of evolution. Moreover, by examining the average hydrophobicity at that and nearby positions, the same alignment might also suggest a pattern of residue solvent accessibility consistent with an α-helix. Taken together, these factors would suggest that the glycine of the original protein adopts α-helical structure, rather than random coil. Several types of methods are used to combine all the available data to form a 3-state prediction, including neural networks, hidden Markov models and support vector machines. Modern prediction methods also provide a confidence score for their predictions at every position. Secondary-structure prediction methods are continuously benchmarked, e.g., in the EVA experiment. Based on ~270 weeks of testing, the most accurate methods at present are PsiPRED, SAM, PORTER, PROF and SABLE. Interestingly, it does not seem to be possible to improve upon these methods by taking a consensus of them [citation needed]. The chief area for improvement appears to be the prediction of β-strands; residues confidently predicted as β-strand are likely to be so, but the methods are apt to overlook some β-strand segments (false negatives). There is likely an upper limit of ~90% prediction accuracy overall, due to the idiosyncrasies of the standard method (DSSP) for assigning secondary-structure classes (helix/strand/coil) to PDB structures, against which the predictions are benchmarked. Accurate secondary-structure prediction is a key element in the prediction of tertiary structure, in all but the simplest (homology modeling) cases. For example, a confidently predicted pattern of six secondary structure elements βαββαβ is the signature of a ferredoxin fold. Nucleic acidsNucleic acids also have secondary structure, most notably single-stranded RNA molecules. RNA secondary structure is generally divided into helices (contiguous base pairs), and various kinds of loops (unpaired nucleotides surrounded by helices). The stem-loop structure in which a base-paired helix ends in a short unpaired loop is extremely common and is a building block for larger structural motifs such as cloverleaf structures, which are four-helix junctions such as those found in transfer RNA. Internal loops (a short series of unpaired bases in a longer paired helix) and bulges (regions in which one strand of a helix has "extra" inserted bases with no counterparts in the opposite strand) are also frequent. Finally, both pseudoknots and base triples are present in RNA (though not DNA). Extremely complex DNA secondary structures have been designed and produced in DNA origami and other products of DNA nanotechnology. Since it is almost entirely base pair-mediated, RNA secondary structure can be said to define which bases are paired in a molecule or complex. However, the traditional Watson-Crick base pair is not the only type of pairing that is permissible in RNA; Hoogsteen base pairing is also common. RNA secondary structure predictionSee also RNA structure One application of bioinformatics uses predicted RNA secondary structures in searching a genome for noncoding but functional forms of RNA. For example, microRNAs have canonical long stem-loop structures interrupted by small internal loops. A general method of calculating probable RNA secondary structure is dynamic programming, although this has the disadvantage that it cannot detect pseudoknots or other cases in which base pairs are not fully nested. More general methods are based on stochastic context-free grammars. A web server that implements a type of dynamic programming is Mfold. For many RNA molecules, the secondary structure is highly important to the correct function of the RNA — often more so than the actual sequence. This fact aids in the analysis of non-coding RNA sometimes termed "RNA genes". RNA secondary structure can be predicted with some accuracy by computer (for example, the RNAsoft web server), and many bioinformatics applications use some notion of secondary structure in analysis of RNA. AlignmentBoth protein and RNA secondary structures can be used to aid in multiple sequence alignment. These alignments can be made more accurate by the inclusion of secondary structure information in addition to simple sequence information. This is sometimes less useful in RNA because base pairing is much more highly conserved than sequence. Distant relationships between proteins whose primary structures are unalignable can sometimes be found by secondary structure. See also
References
|
|||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Secondary_structure". A list of authors is available in Wikipedia. | |||||||||||||||||||||||||||||



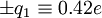 to the carbonyl carbon and oxygen, respectively, and charges of
to the carbonyl carbon and oxygen, respectively, and charges of 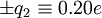 to the amide nitrogen and hydrogen, respectively. The electrostatic energy is
to the amide nitrogen and hydrogen, respectively. The electrostatic energy is
![E = q_{1} q_{2} \left[ \frac{1}{r_{ON}} + \frac{1}{r_{CH}} - \frac{1}{r_{OH}} - \frac{1}{r_{CN}} \right] \cdot 332 \ \mathrm{kcal/mol}](images/math/a/9/e/a9e515ff6849469567acb6d74619726c.png)


