To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Random coil
A random coil is a polymer conformation where the monomer subunits are oriented randomly while still being bonded to adjacent units. It is not one specific shape, but a statistical distribution of shapes for all the chains in a population of macromolecules. The conformation's name is derived from the idea that, in the absence of specific, stabilizing interactions, a polymer backbone will "sample" all possible conformations randomly. Many linear, unbranched homopolymers — in solution, or above their melting temperatures — assume (approximate) random coils. Even copolymers with monomers of unequal length will distribute in random coils if the subunits lack any specific interactions. The parts of branched polymers may also assume random coils. Below their melting temperatures, most thermoplastic polymers (polyethylene, nylon, etc.) have amorphous regions in which the chains approximate random coils, alternating with regions which are crystalline. The amorphous regions contribute elasticity and the crystalline regions contribute strength and rigidity. More complex polymers such as proteins, with various interacting chemical groups attached to their backbones, self-assemble into well-defined structures. But segments of proteins, and polypeptides that lack secondary structure, are often assumed to exhibit a random coil conformation in which the only fixed relationship is the joining of adjacent amino acid residues by a peptide bond. This is not actually the case, since the ensemble will be energy weighted due to interactions between amino acid side chains, with lower-energy conformations being present more frequently. In addition, even arbitrary sequences of amino acids tend to exhibit some hydrogen bonding and secondary structure. For this reason, the term "statistical coil" is occasionally preferred. The conformational entropy associated with the random coil state significantly contributes to its energetic stabilization and accounts for much of the energy barrier to protein folding. A random coil conformation can be detected using spectroscopic techniques. The arrangement of the planar amide bonds results in a distinctive signal in circular dichroism. The chemical shift of amino acids in a random coil conformation is well known in nuclear magnetic resonance (NMR). Deviations from these signatures often indicates the presence of some secondary structure, rather than complete random coil. Furthermore, there are signals in multidimensional NMR experiments which indicate that stable, non-local amino acid interactions are absent for polypeptides in a random coil conformation. Likewise, in the images produced by crystallography experiments, segments of random coil simply result in a reduction in "electron density" or contrast. A randomly coiled state for any polypeptide chain can be attained by denaturing the system. However, there is evidence that proteins are perhaps never truly random coils, even when denatured (Shortle & Ackerman). Product highlight
Random walk model: The Gaussian chain
There are an enormous number of different ways in which a chain can be curled around in a relatively compact shape, like an unraveling ball of twine with lots of open space, and comparatively few ways it can be more or less stretched out. So if each conformation has an equal probability or statistical weight, chains are much more likely to be ball-like than they are to be extended — a purely entropic effect. In an ensemble of chains, most of them will therefore be loosely balled up. Or, equivalently, this is the kind of shape any one of them will have most of the time. Consider a linear polymer to be a freely-jointed chain with N subunits, each of length The average (root mean square) end-to-end distance for the chain, Note that although this model is termed a "Gaussian chain", the distribution function is not a gaussian (normal) distribution. The end-to-end distance probability distribution function of a Gaussian chain is non-zero only for r > 0. [1] Real polymersA real polymer is not freely-jointed. A -C-C- single bond has a fixed tetrahedral angle of 109.5 degrees. The value of L is well-defined for, say, a fully extended polyethylene or nylon, but it is less than N x l because of the zig-zag backbone. There is, however, free rotation about many chain bonds. The model above can be enhanced. A longer, "effective" unit length can be defined such that the chain can be regarded as freely-jointed, along with a smaller N, such that the constraint L = N x l is still obeyed. It, too, gives a Gaussian distribution. However, specific cases can also be precisely calculated. The average end-to-end distance for freely-rotating (not freely-jointed) polymethylene (polyethylene with each -C-C- considered as a subunit) is l times the square root of 2N, an increase by a factor of about 1.4. Unlike the zero volume assumed in a random walk calculation, all real polymers' segments occupy space because of the van der Waals radii of their atoms, including bulky substituent groups which interfere with bond rotations. This can also be taken into account in calculations. All such effects increase the mean end-to-end distance. Because their polymerization is stochastically driven, chain lengths in any real population of synthetic polymers will obey a statistical distribution. In that case, we should take N to be an average value. Also, many polymers have random branching. Even with corrections for local constraints, the random walk model ignores steric interference between chains, and between distal parts of the same chain. A chain often can’t move from a given conformation to a closely related one by a small displacement because one part of it would have to pass through another part, or through a neighbor. We may still hope that the ideal-chain, random-coil model will be at least a qualitative indication of the shapes and dimensions of real polymers in solution, and in the amorphous state, so long as there are only weak physicochemical interactions between the monomers. This model, and the Flory-Huggins Solution Theory, for which Paul Flory received the Nobel Prize in Chemistry in 1974, ostensibly apply only to ideal, dilute solutions. But there is reason to believe (e.g., neutron diffraction studies) that excluded volume effects may cancel out, so that under certain conditions, chain dimensions in amorphous polymers have approximately the ideal, calculated size.[1] When separate chains interact cooperatively, notably in forming crystalline regions in solid thermoplastics, a different mathematical approach must be used. Stiffer polymers such as helical polypeptides, Kevlar, and double-stranded DNA can be treated by the worm-like chain model. See alsoExternal links and references
Categories: Polymers | Polymer chemistry | Organic chemistry | Physical chemistry |
|||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Random_coil". A list of authors is available in Wikipedia. |



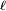 , that occupy zero volume, so that no part of the chain excludes another from any location. One can regard the segments of each such chain in an ensemble as performing a random walk (or "random flight") in three dimensions, limited only by the constraint that each segment must be joined to its neighbors. This is the
, that occupy zero volume, so that no part of the chain excludes another from any location. One can regard the segments of each such chain in an ensemble as performing a random walk (or "random flight") in three dimensions, limited only by the constraint that each segment must be joined to its neighbors. This is the 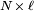 . If we assume that each possible chain conformation has an equal statistical weight, it can be
. If we assume that each possible chain conformation has an equal statistical weight, it can be 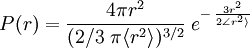
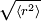 , turns out to be
, turns out to be 

