To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Variational Monte CarloVariational Monte Carlo(VMC) is a quantum Monte Carlo method that applies the variational method to approximate the ground state of the system. Product highlightThe expectation value necessary can be written in the x representation as
Following the Monte Carlo method for evaluating integrals, we can interpret VMC is no different from any other variational method, except that since the many-dimensional integrals are evaluated numerically, we only need to calculate the value of the possibly very complicated wave function, which gives a large amount of flexibility to the method. One of the largest gains in accuracy over writing the wave function separably comes from the introduction of the so-called Jastrow factor, where the wave function is written as Wave Function Optimization in VMCQMC calculations crucially depend on the quality of the trial-function, and so it is essential to have an optimized wave-function as close as possible to the ground state. The problem of function optimization is a very important research topic in numerical simulation. In QMC, in addition to the usual difficulties to find the minimum of multidimensional parametric function, the statistical noise is present in the estimate of the cost function (usually the energy), and its derivatives , required for an efficient optimization. Different cost functions and different strategies were used to optimize a many-body trial-function. Usually three cost functions were used in QMC optimization energy, variance or a linear combination of them. In this thesis we always used energy optimization. The variance optimization have the advantage to be bounded by below, to be positive defined and its minimum is known, but different authors recently showed that the energy optimization is more effective than the variance one. There are different motivations for this: first, usually one is interested in the lowest energy rather than in the lowest variance in both variational and diffusion Monte Carlo; second, variance optimization takes many iterations to optimize determinant parameters and often the optimization can get stuck in multiple local minimum and it suffers of the "false convergence" problem; third energy-minimized wave functions on average yield more accurate values of other expectation values than variance minimized wave functions do. The optimization strategies can be divided into three categories. The first strategy is based on correlated sampling together with deterministic optimization methods. Even if this idea yielded very accurate results for the first-row atoms, this procedure can have problems if parameters affect the nodes, and moreover density ratio of the current and initial trial-function increases exponentially with the size of the system. In the second strategy one use a large bin to evaluate the cost function and its derivatives in such way that the noise can be neglected and deterministic methods can be used. The third approach, is based on an iterative technique to handle directly with noise functions. The first example of these methods is the so called Stochastic Gradient Approximation (SGA), that was used also for structure optimization. Recently an improved and faster approach of this kind was proposed the so called Stochastic Reconfiguration (SR) method. References
Categories: Quantum chemistry | Monte Carlo methods |
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Variational_Monte_Carlo". A list of authors is available in Wikipedia. |



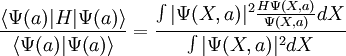 .
.
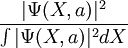 as a probability distribution function, sample it, and evaluate the energy expectation value
as a probability distribution function, sample it, and evaluate the energy expectation value 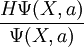 , and minimize
, and minimize 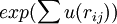 , where
, where 

