L'intelligence artificielle analyse les spectres chimiques en quelques minutes
Des chercheurs de l'université d'Iéna, du Helmholtz-Zentrum Berlin et de Zakodium développent un système d'intelligence artificielle destiné à l'analyse des structures chimiques

Annonces


Déterminer quelle substance a effectivement été produite dans un tube à essai ou un ballon de chimie est l’une des tâches centrales de la chimie. Cependant, notamment dans le cas de composés complexes ou inédits, cela peut s’avérer extrêmement chronophage, même pour des spécialistes expérimentés. Une équipe de recherche de l’université Friedrich Schiller de Iéna, du Centre Helmholtz de Berlin pour les matériaux et l’énergie, de l’Institut Helmholtz des polymères pour les applications énergétiques de Iéna et de la société suisse de logiciels Zakodium Sárl a désormais mis au point un système d’intelligence artificielle (IA) qui propose des structures moléculaires appropriées à partir des données brutes issues de mesures spectroscopiques et évalue leur plausibilité. Ce système est librement accessible et a été présenté dans la revue Nature Communications.
Pourquoi la détermination de la structure est-elle si difficile ?
« Quiconque synthétise une molécule doit également prouver sa structure chimique », explique le Dr Kevin Jablonka de l’université d’Iéna. Il ajoute : « Pour ce faire, les chimistes ont généralement recours à des techniques analytiques telles que la spectroscopie par résonance magnétique nucléaire (RMN), la spectroscopie infrarouge et la spectrométrie de masse. Chacune de ces méthodes fournit des indices sur la structure, mais souvent de manière limitée. Les nombreux signaux de mesure individuels forment donc une sorte de puzzle chimique qu’il faut résoudre correctement. »
L’analyse de la structure s’avère souvent particulièrement difficile pour les nouvelles molécules qui n’ont jamais été décrites auparavant, notamment parce que les données de mesure obtenues dans la pratique sont fréquemment loin d’être idéales. « Les impuretés présentes dans une substance peuvent générer leurs propres signaux ou se superposer à ceux du composé cible », explique Jablonka. « C’est là que notre système présente un atout particulier : pour les spectres RMN protoniques, qui sont couramment mesurés très fréquemment, il est capable de gérer les impuretés présentes dans les échantillons réels. »
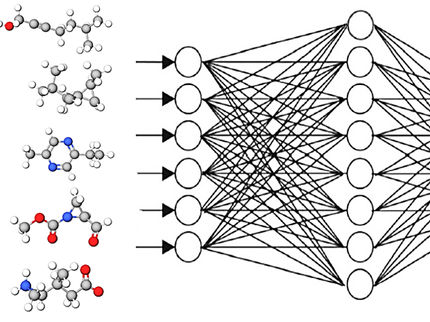
Comment fonctionne le SECS
« Le nouveau système, SECS, combine deux approches d’intelligence artificielle », explique Adrian Mirza, premier auteur de l’étude. « Tout d’abord, le modèle apprend à traduire les spectres et les structures moléculaires en une représentation mathématique commune. Un algorithme évolutif affine ensuite les résultats en modifiant pas à pas les molécules candidates, en ajoutant ou en supprimant des atomes et des liaisons, et en vérifiant à plusieurs reprises si le résultat correspond mieux aux données de mesure. »
Le système produit finalement une liste classée des structures possibles, accompagnée de scores de similarité basés sur le contexte chimique.
Comparable à des spécialistes expérimentés
« Lors d’un test comparatif impliquant différentes méthodes spectroscopiques, SECS a identifié la structure moléculaire correcte comme sa prédiction la mieux classée dans plus de 80 % des cas », précise Jablonka, décrivant les performances du système. Celui-ci s’est également révélé capable d’égaler les experts humains lors d’une comparaison directe. « Dans le cadre d’une étude pilote, nous avons demandé à des chimistes de résoudre 20 problèmes complexes de détermination de structure par RMN », explique M. Jablonka. Résultat : l’IA a atteint un niveau de performance comparable à celui des spécialistes participants.
« Cependant, nous ne considérons pas SECS comme un substitut à l’expertise humaine », souligne M. Mirza. « Le système peut fournir un deuxième avis très utile. » Si les structures proposées sont plausibles et obtiennent des scores élevés, cela renforce la confiance dans l’interprétation. « Si, en revanche, les structures suggérées diffèrent considérablement de la structure moléculaire attendue, il peut être utile d’y jeter un œil de plus près », ajoute Jablonka.
Un outil ouvert à la recherche
Le code source, les données du modèle et une version d’essai de l’application sont accessibles au public. D’après les informations fournies lors d’échanges avec les chercheurs, la version web actuelle est principalement conçue pour l’évaluation directe de données brutes de RMN protonique unidimensionnelle. La prise en charge d’autres types de spectres et d’ensembles de données brutes plus complexes est prévue pour les prochaines versions.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale
Autres actualités du département science


















































