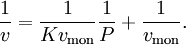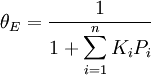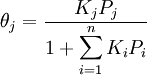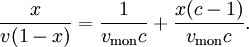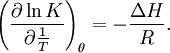To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
AdsorptionAdsorption is a process that occurs when a gas or liquid solute accumulates on the surface of a solid or a liquid (adsorbent), forming a molecular or atomic film (the adsorbate). It is different from absorption, in which a substance diffuses into a liquid or solid to form a solution. The term sorption encompasses both processes, while desorption is the reverse process. Adsorption is operative in most natural physical, biological, and chemical systems, and is widely used in industrial applications such as activated charcoal, synthetic resins and water purification. Adsorption, ion exchange and chromatography are sorption processes in which certain adsorptives are selectively transferred from the fluid phase to the surface of insoluble, rigid particles suspended in a vessel or packed in a column. Similar to surface tension, adsorption is a consequence of surface energy. In a bulk material, all the bonding requirements (be they ionic, covalent or metallic) of the constituent atoms of the material are filled. But atoms on the (clean) surface experience a bond deficiency, because they are not wholly surrounded by other atoms. Thus it is energetically favourable for them to bond with whatever happens to be available. The exact nature of the bonding depends on the details of the species involved, but the adsorbed material is generally classified as exhibiting physisorption or chemisorption. Product highlight
Adsorption isothermsAdsorption is usually described through isotherms, that is, functions which connect the amount of adsorbate on the adsorbent, with its pressure (if gas) or concentration (if liquid). The first isotherm is due to Freundlich and Küster (1894) and it is a purely empirical formula valid for gaseous adsorbates: Langmuir isothermIn 1916, Irving Langmuir published a new isotherm for gases adsorbed on solids, which retained his name. It is an empirical isotherm derived from a proposed kinetic mechanism. It is based on four hypotheses:
These four points are seldom true: there are always imperfections on the surface, adsorbed molecules are not necessarily inert, the mechanism is clearly not the same for the very first molecules as for the last to adsorb. The fourth condition is the most troublesome, as often more molecules can adsorb on the monolayer, but this problem is solved by the BET isotherm. Langmuir suggests that adsorption takes place through this mechanism: A(g) + S ⇌ AS, where A is a gas molecule and S is an adsorption site. The direct and inverse rate constants are k and k-1. If we define surface coverage, θ, as the fraction of the adsorption sites occupied, in the equilibrium we have
For very low pressures θ is difficult to measure experimentally; usually, the adsorbate is a gas and the adsorbed quantity is given at standard temperature and pressure (STP) volume per gram of adsorbent. Therefore, if we call vmon the STP volume of adsorbate required to form a monolayer on the adsorbent (per gram of adsorbent too), Through its slope and y-intercept we can obtain vmon and K, which are constants for each adsorbent/adsorbate pair at a given temperature. vmon is related to the number of adsorption sites through the ideal gas law. If we assume that the number of sites is just the whole area of the solid divided into the cross section of the adsorbate molecules, we can easily calculate the surface area of the adsorbent. Surface area of adsorbents depends on their structure, the more pores they have, the greater the area, which has a big influence on reactions on surfaces. If more than one gas adsorbs on the surface, we call θE the fraction of empty sites and we have and where i is each one of the gases that adsorb. Frumkin isothermFrumkin isotherm is an extension of Langmuir isotherm. It states that adsorbed molecules do interact and affect further adsorption by either repulsion or attraction of molecules.
BET isothermOften molecules do form multilayers, that is, some are adsorbed on already adsorbed molecules and the Langmuir isotherm is not valid. In 1938 Stephan Brunauer, Paul Emmett and Edward Teller developed an isotherm that takes into account that possibility. The proposed mechanism is now:
The derivation of the formula is more complicated than Langmuir's (see links for complete derivation). We obtain: x is the pressure divided into the vapour pressure for the adsorbate at that temperature, v is the STP volume of adsorbed adsorbate, vmon is the STP volume of the amount of adsorbate required to form a monolayer and c is the equilibrium constant K we used in Langmuir isotherm multiplied by the vapour pressure of the adsorbate. The biggest step in BET isotherm is to consider that the successive equilibria for all the layers except for the first are equal to the liquefaction of the adsorbate. Langmuir isotherm is usually better for chemisorption and BET isotherm works better for physisorption. Adsorption enthalpyAdsorption constants are equilibrium constants, therefore they obey van 't Hoff's equation: As can be seen in the formula, the variation of K must be isosteric, that is, at constant coverage. If we start from BET isotherm and assume that the entropy change is the same for liquefaction and adsorption we obtain ΔHads = ΔHliq − RTlnc, that is to say, adsorption is more exothermic than liquefaction. AdsorbentsCharacteristics and general requirements
The adsorbents are used usually in the form of spherical pellets, rods, moldings or monoliths with hydrodynamic diameter between 0.5 and 10 mm. They must have high abrasion resistance, high thermal stability and small micropore diameter, which results in higher exposed surface area and hence high capacity of adsorption. The adsorbents must also have a distinct macropore structure which enables fast transport of the gaseous vapours. Different types of industrial adsorbents generally fall into three classes:
Silica gelSilica gel is a chemically inert, nontoxic, polar and dimensionally stable (< 400 °C) amorphous form of SiO2. It is prepared by the reaction between sodium silicate and sulphuric acid, which is followed by a series of after-treatment processes such as aging, pickling, etc. These after treatment methods results in various pore size distributions on its surface. Silica is also used for drying of process air (e.g. oxygen, natural gas etc) and adsorption of higher (polar) hydrocarbons from natural gas. ZeolitesZeolites are natural or synthetic aluminum silicates which form a regular crystal lattice and release water at high temperature. Zeolites are polar in nature. They are manufactured by hydrothermal synthesis of sodium aluminosilicate in an autoclave followed by ion exchange with certain cations (Na+, Li+, Ca++, K+). The channel diameter of zeolite cages usually ranges from 2 to 9 Å (200 to 900 pm). This process is followed by drying of microcrystals, which are palletized with a binder, to form macropores and thermally activated at a temperature of 6500 °C. Zeolites are applied in drying of process air (only traces), CO2 removal from natural gas, CO removal from reforming gas and air separation. Non-polar zeolites are synthesized by dealumination of polar zeolites. This is done by treating the zeolite with steam at elevated temperatures, greater than 500 °C (1000 °F). This high temperature heat treatment breaks the aluminum-oxygen bonds and the aluminum atom is expelled from the zeolite framework. Non-polar zeolites are mostly used in non-polar organics removal. Activated carbonThey are highly porous, amorphous solids consisting of microcrystallites with a graphite lattice. They are non-polar and cheap. One of their main drawbacks is that they are combustible. Activated carbon can be manufactured from carbonaceous material, including coal (bituminous, subbituminous, and lignite), peat, wood, or nutshells (i.e., coconut). The manufacturing process consists of two phases, carbonization and activation. The carbonization process includes drying and then heating to separate by-products, including tars and other hydrocarbons, from the raw material, as well as to drive off any gases generated. The carbonization process is completed by heating the material at 400–600 °C in an oxygen-deficient atmosphere that cannot support combustion. The carbonized particles are “activated” by exposing them to an oxidizing agent, usually steam or carbon dioxide at high temperature. This agent burns off the pore blocking structures created during the carbonization phase and so, they develop a porous, three-dimensional graphite lattice structure. The size of the pores developed during activation is a function of the time that they treated in this stage. Longer exposure times result in larger pore sizes. The most popular aqueous phase carbons are bituminous based because of their hardness, abrasion resistance, pore size distribution, and low cost, but their effectiveness needs to be tested in each application to determine the optimal product. Activated carbon is used for adsorption of organic substances and non-polar adsorptives and it is also usually used for waste gas (and waste water) treatment. It is the most widely used adsorbent. Its usefulness derives mainly from its large micropore and mesopore volumes and the resulting high surface area. Portal site mediated adsorptionPortal site mediated adsorption is a model for site-selective activated gas adsorption in metallic catalytic systems which contain a variety of different adsorption sites. In such systems, low-coordination "edge and corner" defect-like sites can exhibit significantly lower adsorption enthalpies than high-coordination (basal plane) sites. As a result, these sites can serve as "portals" for very rapid adsorption to the rest of the surface. The phenomena relies on the common "spillover" effect, where certain adsorbed species exhibit high mobility on some surfaces. The model explains seemingly inconsistent observations of gas adsorption thermodynamics and kinetics in catalytic systems where surfaces can exist in a range of coordination structures, and it has been successfully applied to bimetallic catalytic systems where synergistic activity is observed. The original model was developed by King and co-workers (Narayan et al. 1998 and VanderWiel et al. 1999) to describe hydrogen adsorption on silica-supported silver-ruthenium and copper-ruthenium bimetallic catalysts. The same group applied the model to CO hydrogenation (Fischer-Tropsch synthesis). Zupanc et al. (2002) subsequently confirmed the same model on magnesia-supported cesium-ruthenium bimetallic catalysts. Adsorption in virusesAdsorption is the first step in the viral infection cycle. The next steps are penetration, uncoating, synthesis (transcription if needed, and translation), and release. The virus replication cycle is similar, if not the same, for all types of viruses. Factors such as transcription may or may not be needed if the virus is able to integrate its genomic information in the cell's nucleus, or if the virus can replicate itself directly within the cell's cytoplasm. References
See also
Categories: Surface chemistry | Materials science | Chemical engineering | Colloidal chemistry |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Adsorption". A list of authors is available in Wikipedia. |



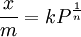 , where x is the adsorbed quantity, m is the mass of adsorbent, P is the pressure of adsorbate and k and n are empirical constants for each adsorbent-adsorbate pair at each temperature. The function has an asymptotic maximum. As the temperature increases, the adsorbed quantity rises more slowly and more pressure is required to achieve the maximum.
, where x is the adsorbed quantity, m is the mass of adsorbent, P is the pressure of adsorbate and k and n are empirical constants for each adsorbent-adsorbate pair at each temperature. The function has an asymptotic maximum. As the temperature increases, the adsorbed quantity rises more slowly and more pressure is required to achieve the maximum.
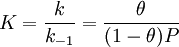 or
or 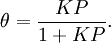
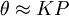 and for high pressures
and for high pressures 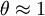
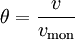 and we obtain an expression for a straight line:
and we obtain an expression for a straight line: