To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
GF methodWilson's GF method, sometimes referred to as FG method, is a classical mechanical method to obtain certain internal coordinates for a vibrating semi-rigid molecule, the so-called normal coordinates Qk. Normal coordinates decouple the classical vibrational motions of the molecule and thus give an easy route to obtaining vibrational amplitudes of the atoms as a function of time. In Wilson's GF method it is assumed that the molecular kinetic energy consists only of harmonic vibrations of the atoms, i.e., overall rotational and translational energy is ignored. Normal coordinates appear also in a quantum mechanical description of the vibrational motions of the molecule and the Coriolis coupling between rotations and vibrations. It follows from application of the Eckart conditions that the matrix G-1 gives the kinetic energy in terms of arbitrary linear internal coordinates, while F represents the (harmonic) potential energy in terms of these coordinates. The GF method gives the linear transformation from general internal coordinates to the special set of normal coordinates. Product highlight
The GF methodA non-linear molecule consisting of N atoms has 3N-6 internal degrees of freedom, because positioning a molecule in three-dimensional space requires three degrees of freedom and the description of its orientation in space requires another three degree of freedom. These degrees of freedom must be subtracted from the 3N degrees of freedom of a system of N particles. The atoms in a molecule are bound by a potential energy surface (PES)—also known as force field—which is a function of 3N-6 coordinates. The internal degrees of freedom q1, ..., q3N-6 describing the PES in an optimum way are often non-linear; they are for instance valence coordinates, such as bending and torsion angles and bond stretches. It is possible to write the quantum mechanical kinetic energy operator for such curvilinear coordinates, but it is hard to formulate a general theory applicable to any molecule. This is why Wilson[1] linearized the internal coordinates by assuming small displacements. The linearized version of the internal coordinate qt is denoted by St. The PES V can be Taylor expanded around its minimum in terms of the St. The third term (the Hessian of V) evaluated in the minimum is a force derivative matrix F. In the harmonic approximation the Taylor series is ended after this term. The second term, containing first derivatives, is zero because it is evaluated in the miminum of V. The first term can be included in the zero of energy. Thus,
The classical vibrational kinetic energy has the form: where gst is an element of the metric tensor of the internal (curvilinear) coordinates. The dots indicate time derivatives. Evaluation of the metric tensor g in the minimum q0 of V gives the positive definite and symmetric matrix G = g(q0)-1. One can solve the following two matrix problems simultaneously since they are equivalent to the generalized eigenvalue problem where Because of the form of the generalized eigenvalue problem, the method is called the GF method, often with the name of its originator attached to it: Wilson's GF method. By matrix transposition in both sides of the equation and using the fact that both G and F are symmetric matrices, as are diagonal matrices, one can recast this equation into a very similar one for FG . This is why the method is also referred to as Wilson's FG method. We introduce the vectors which satisfy the relation Upon use of the results of the generalized eigenvalue equation the energy E = T + V (in the harmonic approximation) of the molecule becomes, The Lagrangian L = T - V is The corresponding Lagrange equations are identical to the Newton equations for a set of uncoupled harmonic oscillators. These ordinary second-order differential equations are easily solved yielding Qt as function of time, see the article on harmonic oscillators. Normal coordinates in terms of Cartesian displacement coordinatesOften the normal coordinates are expressed as linear combinations of Cartesian displacement coordinates.
Let RA be the position vector of nucleus A and RA0
the corresponding equilibrium position. Then
where sAt is known as a Wilson s-vector.
If we put the The actual form of the matrix elements of B can be fairly complicated.
Especially for a torsion angle, which involves 4 atoms, it requires tedious vector algebra to derive the corresponding values of the In summation language: Here D is a 3N-6 x 3N matrix which is given by (i) the linearization of the internal coordinates q (an algebraic process) and (ii) solution of Wilson's GF equations (a numeric process). Relation with Eckart conditionsFrom the invariance of the internal coordinates St under overall rotation and translation of the molecule, follows the same for the linearized coordinates stA. It can be shown that this implies that the following 6 conditions are satisfied by the internal coordinates, These conditions follow from the Eckart conditions that hold for the displacement vectors, See this article for more details. ReferencesCited reference
Further references
Categories: Spectroscopy | Molecular physics | Quantum chemistry |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "GF_method". A list of authors is available in Wikipedia. |



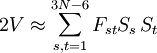 .
.
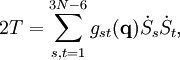
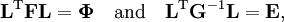
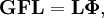
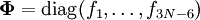 and
and 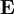 is the unit matrix. The matrix L-1 contains the normal coordinates Qk in its rows:
is the unit matrix. The matrix L-1 contains the normal coordinates Qk in its rows:
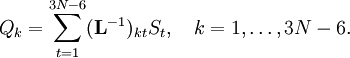
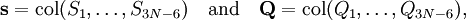
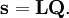
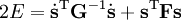
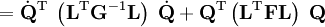
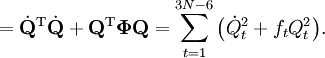
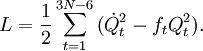
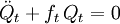
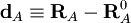 is by definition the Cartesian displacement coordinate of nucleus A.
Wilson's linearizing of the internal curvilinear coordinates qt expresses the coordinate St in terms of the displacement coordinates
is by definition the Cartesian displacement coordinate of nucleus A.
Wilson's linearizing of the internal curvilinear coordinates qt expresses the coordinate St in terms of the displacement coordinates
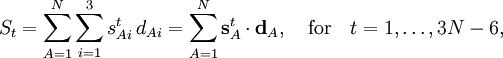
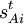 into a 3N-6 x 3N matrix B, this equation becomes in matrix language
into a 3N-6 x 3N matrix B, this equation becomes in matrix language
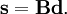
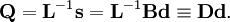
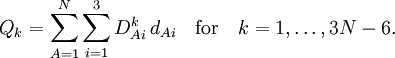
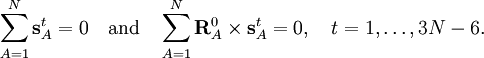
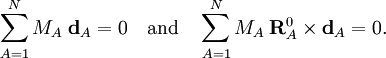
Last viewed


