To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Hybrid functionalA hybrid functional is an exchange-correlation functional used in density functional theory (DFT) that incorporates a portion of exact exchange from Hartree-Fock theory with exchange and correlation from other sources (ab initio, such as LDA, or empirical). Product highlight
OriginThe hybrid approach to density functionals was first introduced by Axel Becke in 1993[1]. Hybridization with Hartree-Fock (exact) exchange provides a simple scheme for improving many molecular properties, such as atomization energies, bond lengths and vibration frequencies, which tend to be poorly described with simple ab initio functionals.[2] MethodThe exchange-correlation functional for a hybrid is usually a linear combination of the Hartree-Fock exchange (
List of hybrid functionals (in Gaussian 03)
See: G03 Manual: DFT. References
|
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Hybrid_functional". A list of authors is available in Wikipedia. |



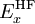 ) and some other one or combination of exchange and correlation functionals. The parameter(s) relating the amount of each functional can be arbitrarily assigned and is usually fitted to reproduce well some set of observables (bond lengths,
) and some other one or combination of exchange and correlation functionals. The parameter(s) relating the amount of each functional can be arbitrarily assigned and is usually fitted to reproduce well some set of observables (bond lengths, 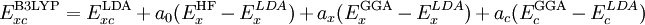
 ,
, 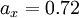 , and
, and 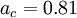 are the three empirical parameters;
are the three empirical parameters; 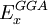 and
and 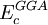 are the generalized gradient approximation formulated with the Becke 88 exchange functional
are the generalized gradient approximation formulated with the Becke 88 exchange functional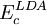 the VWN correlation functional (see
the VWN correlation functional (see 

