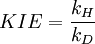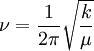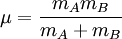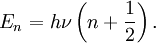To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Kinetic isotope effectThe kinetic isotope effect (KIE) is a variation in the reaction rate of a chemical reaction when an atom in one of the reactants is replaced by one of its isotopes. It is also called isotope fractionation, although this term is technically somewhat broader in meaning. A KIE involving hydrogen and deuterium is represented as: with kH and kD reaction rate constants. An isotopic substitution will greatly modify the reaction rate when the isotopic replacement is in a chemical bond that is broken or formed in the rate limiting step. In such a case, the rate change is termed a primary isotope effect. When the substitution is not involved in the bond that is breaking or forming, one may still observe a smaller rate change, termed a secondary isotope effect. Thus, the magnitude of the kinetic isotope effect can be used to elucidate the reaction mechanism. If other steps are partially rate-determining, the effect of isotopic substitution will be masked. Isotopic rate changes are most pronounced when the relative mass change is greatest. For instance, changing a hydrogen atom to deuterium represents a 100% increase in mass, whereas in replacing carbon-12 with carbon-13, the mass increases by only 8%. The rate of a reaction involving a C-H bond is typically 6 to 10 times faster than the corresponding C-D bond, whereas a 12C reaction is only ~1.04 times faster than the corresponding 13C reaction (even though, in both cases, the isotope is one atomic mass unit heavier). Isotopic substitution can modify the rate of reaction in a variety of ways. In many cases, the rate difference can be rationalized by noting that the mass of an atom affects the vibration frequency of the chemical bond that it forms, even if the electron configuration is nearly identical. Heavier atoms will (classically) lead to lower vibration frequencies, or, viewed quantum mechanically, will have lower zero-point energy. With a lower zero-point energy, more energy must be supplied to break the bond, resulting in a higher activation energy for bond cleavage, which in turn lowers the measured rate (see, for example, the Arrhenius equation). The Swain equation relates the kinetic isotope effect for the proton/tritium combination with that of the proton/deuterium combination. Product highlight
Mathematical details in a diatomic moleculeOne approach to studying the effect is for that of a diatomic molecule. The fundamental vibrational frequency (ν) of a chemical bond between atom A and B is, when approximated by a harmonic oscillator: where k is the spring constant for the bond, and μ is the reduced mass of the A-B system: (mi is the mass of atom i). Quantum mechanically, the energy of the n-th level of a harmonic oscillator is given by: Thus, the zero-point energy (n = 0) will decrease as the reduced mass increases. With a lower zero-point energy, more energy is needed to overcome the activation energy for bond cleavage. In changing a carbon-hydrogen bond to a carbon-deuterium bond, k remains unchanged, but the reduced mass µ is different. As a good approximation, on going from C-H to C-D, the reduced mass increases by a factor of approximately 2. Thus, the frequency for a C-D bond should be approximately 1/√2 or 0.71 times that of the corresponding C-H bond. Still, this is a much larger effect than changing the carbon-12 to carbon-13. The secondary Isotope effectThe secondary kinetic isotope effect (SKIE) arises when an isotopic substitution that is not involved in the reaction influences the internal vibrations of the system that via changes in the zero point energy (ZPE) affect the rates of chemical reactions [1]. Such effects are expressed as ratios of rate for the light isotope to that of the heavy isotope and can be "normal" (ratio is greater than or equal to 1) or "inverse" (ratio is less than 1) effects.[2] SKIE are defined as α,β (etc.) secondary isotope effects where such prefixes refer to the position of the isotopic substitution relative to the reaction center.[3] The prefix α refers to the isotope associated with the reaction center while the prefix β refers to the isotope associated with an atom neighboring the reaction center and so on. In physical organic chemistry SKIE is discussed in terms of electronic effects such as induction, bond hybridization or hyperconjugation[4]. These properties are determined by electron distribution, and depend upon vibrationally averaged bond length and angles that are not greatly affected by isotopic substitution. Thus, the use of the term "electronic isotope effect" while legitimate is discouraged from use as it incorrectly suggests that the isotope effect is electronic in nature rather than vibrational.[3] SKIE detection in physical organic chemistry can be explained in terms of changes in orbital hybridisation. When a carbon atom changes from sp3 to sp2 hybridization a number of vibrational modes (stretches, in-plane and out-of-plane bending) undergo large changes. The in-plane and out-of-plane bending in an sp3 hybridized carbon are similar in frequency due to the symmetry of an sp3 hybridized carbon. In an sp2 hybridized carbon the in-plane bend is much stiffer than the out-of-plane bending resulting in a large difference in the frequency, the ZPE and thus the SKIE (which exists when there is a difference in the ZPE of the reactant and transition state).[5] The theoretical maximum change caused by the bending frequency difference has been calculated as 1.4.[5] When undergoing a reaction that changes the hybridization of carbon from sp3 to sp2 the force constant at the transition state is weaker as it is developing sp2 character and a "normal" SKIE is observed with typical values of 1.1 to 1.2.[5] Similarly when going from sp2 to sp3 character the force constants at the tranistion state increase and an inverse SKIE is observered with typical values of 0.8 to 0.9.[5] More generally the SKIE for reversible reactions can be "normal" one way and "inverse" the other if bonding in the transition state is midway in stiffness between substrate and product, or they can be "normal" both ways if bonding is weaker in the transition state, or "inverse" both ways if bonding is stronger in the transition state than in either reactant.[2] An example of an "inverse" α secondary kinetic isotope effect can be seen in the work of Fitzpatrick and Kurtz who used such an effect to distinguish between two proposed pathways for the reaction of d-amino acid oxidase with nitroalkane anions.[6] Path A involved a neucleophilic attack while path B involves a free-radical intermediate. As path A results in the intermediate carbon changing hybridization from sp2 to sp3 an "inverse" a SKIE is expected. If path B occurs then no SKIE should be observed as the free radical intermediate does not change hybridization. An SKIE of 0.84 was observed and Path A verified as shown in the scheme below.
Steric Isotope EffectThe steric isotope effect is an SKIE that does not involve bond breaking or formation. This effect is attributed to the different vibrational amplitudes of isotopologues (molecular entities differing in isotopic composition only.[8])[9] An example of such an effect can be seen in the work of Kurt Mislow et al.[10] who demonstrated through the racemization of 9,10-dihydro-4,5-dimethylphenanthrene that the smaller amplitude of vibration for deuterium as compared to hydrogen in C-H (Carbon -Hydrogen), C-D (Carbon -Deuterium) bonds results in a smaller van der Waals radius or effective size in addition to a difference in the ZPE between the two. When there is a greater effective bulk of molecules containing one over the other this may be manifested by a steric effect on the rate constant. For the example above deuterium racemizes faster than the hydrogen isotopologue resulting in a steric isotope effect.
ApplicationsThe kinetic isotope effect is applied in reaction mechanism elucidation, for instance in the halogenation of toluene:[12] In this particular intramolecular KIE study the radical substitution of hydrogen by bromine is examined with mono-deuterated toluene (obtained by organic reduction of benzyl chloride with zinc and deuterated acetic acid) and N-bromosuccinimide. As hydrogen is replaced by bromine faster than deuterium, the reaction product gets enriched in deuterium. In order to analyze the deuterium composition by means of mass spectroscopy the reaction product is reduced back to toluene with lithium aluminum hydride and a KIE of 4.86 is calculated. This finding is in accordance with the general accepted view of a radical substitution in which a hydrogen atom is removed by a bromine free radical species in the rate-determining step. A large KIE of 5.56 is also reported for reaction of ketones with bromine and sodium hydroxide forming a haloketone with the α-carbonyl positions deuterated.[13] In this reaction the rate-limiting step is enolate formation by proton (deuterium) abstraction from the ketone by base. In this study the KIE is calculated from the reaction rate constants for regular 2,4-dimethyl-3-pentanone and its deuterated isomer by optical density measurements. TunnelingIn some cases, an additional rate enhancement is seen for the lighter isotope, possibly due to quantum mechanical tunnelling. This is typically only observed for hydrogen atoms, which are light enough to exhibit significant tunnelling. This effect has been observed in such reactions as the deprotonation and iodination of nitropropane with hindered pyridine base[14] with a reported KIE of 25 at 25 °C: and in a 1,5-sigmatropic hydrogen shift[15] although it is observed that it is difficult to extrapolate experimental values obtained at elevated temperatures to lower temperatures:[16][17] See also
References
Categories: Chemical kinetics | Physical organic chemistry |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Kinetic_isotope_effect". A list of authors is available in Wikipedia. |