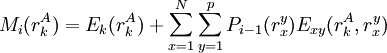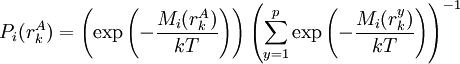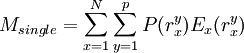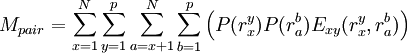To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Self-consistent mean field (biology)The self-consistent mean field (SCMF) method is an adaptation of mean field theory used in protein structure prediction to determine the optimal amino acid side chain packing given a fixed protein backbone. It is faster but less accurate than dead-end elimination and is generally used in situations where the protein of interest is too large for the problem to be tractable by DEE. Product highlight
General principlesLike dead-end elimination, the SCMF method explores conformational space by discretizing the dihedral angles of each side chain into a set of rotamers for each position in the protein sequence. The method iteratively develops a probabilistic description of the relative population of each possible rotamer at each position, and the probability of a given structure is defined as a function of the probabilities of its individual rotamer components. The basic requirements for an effective SCMF implementation are:
The process is generally initialized with a uniform probability distribution over the rotamers - that is, if there are p rotamers at the kth position in the protein, then the probability of any individual rotamer Mean-field energiesThe energy of an individual rotamer rk is dependent on the "mean-field" energy of the other positions - that is, at every other position, each rotamer's energy contribution is proportional to its probability. For a protein of length N with p rotamers per residue, the energy at the current iteration is described by the following expression. Note that for clarity, the mean-field energy at iteration i is denoted by Mi, whereas the precomputed energies are denoted by E, and the probability of a given rotamer is denoted by These mean-field energies are used to update the probabilities through the Boltzmann law: where k is the Boltzmann constant and T is the temperature factor. Energy of the systemAlthough computing the system energy is not required in carrying out the SCMF method, it is useful to know the overall energies of the converged results. The system energy Msys consists of two sums:
where the addends are defined as: ConvergencePerfect convergence for the SCMF method would result in a probability of 1 for exactly one rotamer at each position k in the protein, and a probability of zero for all other rotamers at each position. Convergence to a unique solution requires probabilities close to 1 for exactly one rotamer at each position. In practice, especially when higher temperatures are used, the algorithm instead identifies a small number of high-probability rotamers at each position, allowing the resulting conformations' relative energies to then be enumerated (based on the precomputed energies, not on those derived from the mean-field approximation). One way to improve convergence is to run again at a lower temperature using the probabilities calculated from a previous higher-temperature run. AccuracyUnlike dead-end elimination, SCMF is not guaranteed to converge on the optimal solution. However, it is deterministic (as in, it will converge to the same solution every time given the same initial conditions), unlike alternatives that rely on Monte Carlo analysis. By comparison to DEE, which is guaranteed to find the optimal solution, SCMF is faster but less accurate overall; it is significantly better at identifying correct side chain conformations in the protein's core than it is on identifying correct surface conformations. Geometric packing constraints are less restrictive on the surface and thus provide fewer boundaries to the conformational search. References
|
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Self-consistent_mean_field_(biology)". A list of authors is available in Wikipedia. |



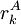 is
is 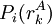 .
.