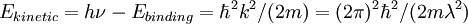To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
XANESX-ray Absorption Near Edge Structure (XANES) is a type of absorption spectroscopy. XANES data indicate the absorption peaks due to the photoabsorption cross section in the X-ray Absorption Spectra (XAS) observed in the energy region, extending over a range of about 100 eV, between the edge region and the EXAFS region. Here the XANES spectroscopy and its applications in these last 26 years is described. XANES is also known as NEXAFS (Near Edge X-ray Absorption Fine Structure) when applied to surface and molecular science and it is described elsewhere. XANES has to be distinguished from edge and EXAFS spectroscopy. The first difference concerns the energy range above the absorption edge. Product highlight
ENERGY RANGEEdge energy rangeIn the absorption edge region of metals, the photoelectron is excited to the first unoccupied level above the Fermi level. Therefore its mean free path in a pure single crystal at zero temperature is as large as infinite, and it remains very large, increasing the energy of the final state up to about 5 eV above the Fermi level. Beyond the role of the unoccupied density of states and matrix elements in single electron excitations, many-body effects appear as an "infrared singularity" at the absorption threshold in metals. In the absorption edge region of insulators the photoelectron is excited to the first unoccupied level above the chemical potential but the unscreened core hole forms a localized bound state called core exciton. EXAFS energy range
The fine structure in the x-ray absorption spectra in the high energy range extending from about 150 eV beyond the ionization potential is a powerful tool to determine the atomic pair distribution (i.e. interatomic distances) with a time scale of about 10-15 s. In fact the final state of the excited photoelectron in the high kinetic energy range (150-2000 eV ) is determined only by single backscattering events due to the low amplitude photoelectron scattering. XANES energy rangeIn the XANES region, starting about 5 eV beyond the absorption threshold, because of the low kinetic energy range (5-150 eV) the photoelectron backscattering amplitude by neighbor atoms is very large so that multiple scattering events become dominant in the XANES spectra. The different energy range between XANES and EXAFS can be also explained in a very simple manner by the comparison between the photoelectron wavelength λ and the interatomic distance of the photoabsorber-backscatterer pair. The photoelectron kinetic energy is connected with the wavelength λ by the following relation: that means that for high energy the wavelength is shorter than interatomic distances and hence the EXAFS region corresponds to a single scattering regime; while for lower E, λ is larger than interatomic distances and the XANES region is associated with a multiple scattering regime. FINAL STATESThe absorption peaks of XANES spectra are determined by multiple scattering resonances of the photoelectron excited at the atomic absorption site and scattered by neighbor atoms. The local character of the final states is determined by the short photoelectron mean free path, that is strongly reduced (down to about 0.3 nm at 50 eV) in this energy range because of inelastic scattering of the photoelectron by electron-hole excitations (excitons) and collective electronic oscillations of the valence electrons called plasmons. ApplicationsMuch chemical information can be extracted from the XANES region: formal valence (very difficult to experimentally determine in a nondestructive way); coordination environment (e.g., octahedral, tetrahedral coordination) and subtle geometrical distortions of it. Transitions to bound vacant states just above the Fermi level can be seen. Thus XANES spectra can be used as a probe of the unoccupied band structure of a material. The near-edge structure is characteristic of an environment and valence state hence one of its more common uses is in fingerprinting: if you have a mixture of sites/compounds in a sample you can fit the measured spectra with a linear combinations of XANES spectra of known species and determine the proportion of each site/compound in the sample. One example of such a use is the determination of the oxidation state of the plutonium in the soil at Rocky Flats. HistoryThe acronym XANES has been first introduced in 1980 to interpret the XANES spectra measured at the Stanford Synchrotron Radiation Laboratory (SSRL) determined by multiple scattering resonances by A. Bianconi. In 1982 the first paper on the application of XANES spectroscopy for determination of the local structural geometrical distortions using multiple scatterial theory was published by the collaboration between A. Bianconi, P. J. Durham, J. B. Pendry. In 1983 the first NEXAFS paper appeared for molecules adsorbed on surfaces. The first XAFS paper appeared in 1987 for the interpretation of the intermediate region between EXAFS and XANES. References
Lectures
Bibliography
Categories: Materials science | Condensed matter physics | Environmental chemistry | Spectroscopy |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "XANES". A list of authors is available in Wikipedia. |