Une nouvelle méthode améliore la précision des potentiels appris par la machine pour la simulation des catalyseurs
Faire le lien entre la précision et l'efficacité dans la conception des catalyseurs
Annonces
Les catalyseurs jouent un rôle indispensable dans la fabrication moderne. Plus de 80 % de tous les produits manufacturés, des produits pharmaceutiques aux plastiques, reposent sur des processus catalytiques à un stade ou à un autre de la production. Les métaux de transition, en particulier, sont des catalyseurs très efficaces car leurs orbitales d partiellement remplies leur permettent d'échanger facilement des électrons avec d'autres molécules. Toutefois, cette propriété même les rend difficiles à modéliser avec précision, ce qui nécessite des descriptions exactes de leur structure électronique.
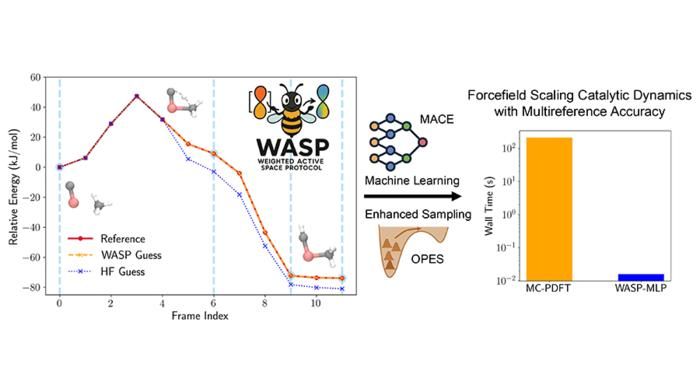
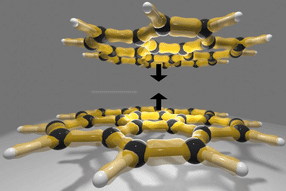
Précision et accélération obtenues avec le Weighted Active Space Protocol (WASP) pour l'activation du méthane sur le carbure de titane.
Figure courtesy of Seal et al.
La conception de catalyseurs à base de métaux de transition efficaces, capables de fonctionner dans des conditions réalistes, exige plus qu'un instantané statique d'une réaction. Nous devons plutôt saisir l'image dynamique - comment les molécules se déplacent et interagissent à différentes températures et pressions, où le mouvement atomique façonne fondamentalement la performance catalytique.
Pour relever ce défi, le laboratoire du professeur Laura Gagliardi de la Pritzker School of Molecular Engineering (UChicago PME) et du département de chimie de l'université de Chicago a mis au point un nouvel outil puissant qui exploite les théories de la structure électronique et l'apprentissage automatique pour simuler la dynamique catalytique des métaux de transition avec précision et rapidité.

"Au cours de la dernière décennie, les potentiels appris par la machine ont considérablement fait progresser la façon dont nous simulons la dynamique moléculaire, en offrant rapidité et évolutivité. Pourtant, capturer avec précision la structure électronique des catalyseurs à base de métaux de transition est resté un défi non résolu. Notre nouvelle méthode comble cette lacune en intégrant des méthodes de chimie quantique multiréférences avec des potentiels appris par la machine, ce qui permet d'obtenir à la fois précision et efficacité". a déclaré M. Gagliardi.
Les résultats ont été publiés dans la revue Proceedings of the National Academy of Sciences.
Accélérer les simulations grâce à l'apprentissage automatique
Au cours de la dernière décennie, le groupe de M. Gagliardi a développé la théorie de la fonctionnelle des paires de configurations multiples (MC-PDFT), une méthode de chimie quantique capable de décrire les structures électroniques complexes des réactions des métaux de transition. Bien que la MC-PDFT offre une grande précision, elle est excessivement lente pour simuler la dynamique des systèmes catalytiques, une étape essentielle pour prédire le comportement réel des catalyseurs dans des conditions réalistes.
Pour relever ce défi, l'équipe s'est tournée vers les potentiels interatomiques appris par machine (ML-potentials), qui peuvent capturer la dynamique moléculaire avec une efficacité remarquable. Les potentiels ML ont été largement appliqués à la science des matériaux, mais jusqu'à présent, ils n'avaient jamais été combinés avec succès à des méthodes multiréférences telles que la MC-PDFT.
La raison réside dans un obstacle de longue date : la cohérence de l'étiquetage. Les modèles d'apprentissage automatique nécessitent des étiquettes de propriétés uniques et fiables, telles que les énergies et les forces dérivées des fonctions d'onde, pour chaque géométrie moléculaire le long d'une voie de réaction. Pour les méthodes de chimie quantique multiréférences, l'attribution de ces étiquettes de manière unique est restée un problème non résolu.
Pour relever ce défi, l'étudiant en doctorat Aniruddha Seal, conseillé conjointement par Gagliardi et le professeur Andrew Ferguson, a développé un nouvel algorithme qui génère des fonctions d'onde cohérentes pour les nouvelles géométries sous la forme d'une combinaison pondérée de fonctions d'onde provenant de structures moléculaires précédemment échantillonnées. Plus une nouvelle géométrie est proche d'une géométrie connue, plus sa fonction d'onde ressemble à celle de la structure connue. Cette approche garantit que chaque point d'une voie de réaction se voit attribuer une fonction d'onde unique et cohérente, ce qui permet d'entraîner avec précision les potentiels ML sur des données multiréférentielles.
"C'est un peu comme mélanger des peintures sur une palette", explique M. Seal. "Si je veux créer une nuance de vert plus proche du bleu, j'utiliserai plus de peinture bleue et juste un peu de jaune. Si je veux une nuance qui penche vers le jaune, l'équilibre s'inverse. Plus ma couleur cible est proche de l'une des peintures de base, plus elle influence le mélange. WASP fonctionne de la même manière : il mélange les informations provenant de structures moléculaires proches, en donnant plus de poids à celles qui sont les plus similaires, afin de créer une prédiction précise pour la nouvelle géométrie".
Cette innovation constitue la base du Weighted Active Space Protocol (WASP), un cadre qui combine la précision de la MC-PDFT et l'efficacité de l'apprentissage automatique, développé grâce à une étroite collaboration avec le groupe Parrinello de l'Institut italien de technologie de Gênes, qui réunit l'expertise en matière de théorie de la structure électronique et de potentiels appris par la machine. WASP permet des accélérations spectaculaires : des simulations avec une précision multiréférentielle qui prenaient des mois peuvent désormais être réalisées en quelques minutes seulement.
Impact : Rapprocher la précision et l'efficacité dans la conception des catalyseurs
En alliant précision et rapidité, WASP ouvre la voie à la conception de catalyseurs capables de résister à des conditions réalistes - hautes températures et hautes pressions. Les métaux de transition sont au cœur d'innombrables processus à grande échelle, mais leur complexité a rendu la conception rationnelle des catalyseurs difficile.
Le procédé Haber-Bosch, où le fer sert de catalyseur pour convertir l'azote et l'hydrogène en ammoniac, en est un excellent exemple. Bien qu'il ait été mis au point il y a plus d'un siècle, ce catalyseur à base de fer domine toujours la production d'ammoniac dans le monde entier. Avec le WASP, les chercheurs disposent désormais des outils nécessaires pour explorer des alternatives susceptibles d'accroître l'efficacité, de réduire les sous-produits et de diminuer le coût environnemental.
Jusqu'à présent, le WASP a été démontré avec succès pour la catalyse activée thermiquement, c'est-à-dire les réactions entraînées par la chaleur. La prochaine étape consiste à adapter la méthode aux réactions activées par la lumière, qui sont essentielles pour la conception de nouveaux photocatalyseurs. Les photocatalyseurs sont extrêmement prometteurs pour les technologies, du traitement de l'eau à la production d'énergie.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale

Autres actualités du département science