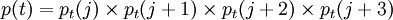To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Chou-Fasman methodThe Chou-Fasman method is an empirical technique for the prediction of secondary structures in proteins, originally developed in the 1970s. [1][2][3] The method is based on analyses of the relative frequencies of each amino acid in alpha helices, beta sheets, and turns based on known protein structures solved with X-ray crystallography. From these frequencies a set of probability parameters were derived for the appearance of each amino acid in each secondary structure type, and these parameters are used to predict the probability that a given sequence of amino acids would form a helix, a beta strand, or a turn in a protein. The method is at most about 50-60% accurate in identifying correct secondary structures, which is significantly less accurate than the GOR method or modern machine learning-based techniques.[4] Product highlightAmino acid propensitiesThe original Chou-Fasman parameters found some strong tendencies among individual amino acids to prefer one type of secondary structure over others. Alanine, glutamate, leucine, and methionine were identified as helix formers, while proline and glycine, due to the unique conformational properties of their peptide bonds, commonly end a helix. The original Chou-Fasman parameters[5] were derived from a very small and non-representative sample of protein structures due to the small number of such structures that were known at the time of their original work. These original parameters have since been shown to be unreliable[6] and have been updated from a current dataset, along with modifications to the initial algorithm.[7] The Chou-Fasman method takes into account only the probability that each individual amino acid will appear in a helix, strand, or turn. Unlike the more complex GOR method, it does not reflect the conditional probabilities of an amino acid to form a particular secondary structure given that its neighbors already possess that structure. This lack of cooperativity increases its computational efficiency but decreases its accuracy, since the propensities of individual amino acids are often not strong enough to render a definitive prediction.[4] AlgorithmThe Chou-Fasman method predicts helices and strands in a similar fashion, first searching linearly through the sequence for a "nucleation" region of high helix or strand probability and then extending the region until a subsequent four-residue window carries a probability of less than 1. As originally described, four out of any six contiguous amino acids were sufficient to nucleate helix, and three out of any contiguous five were sufficient for a sheet. The probability thresholds for helix and strand nucleations are constant but not necessarily equal; originally 1.03 was set as the helix cutoff and 1.00 for the strand cutoff. Turns are also evaluated in four-residue windows, but are calculated using a multi-step procedure because many turn regions contain amino acids that could also appear in helix or sheet regions. Four-residue turns also have their own characteristic amino acids; proline and glycine are both common in turns. A turn is predicted only if the turn probability is greater than the helix or sheet probabilities and a probability value based on the positions of particular amino acids in the turn exceeds a predetermined threshold. The turn probability p(t) is determined as: where j is the position of the amino acid in the four-residue window. If p(t) exceeds an arbitrary cutoff value (originally 7.5e-3), the mean of the p(j)'s exceeds 1, and p(t) exceeds the alpha helix and beta sheet probabilities for that window, then a turn is predicted. If the first two conditions are met but the probability of a beta sheet p(b) exceeds p(t), then a sheet is predicted instead. References
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Chou-Fasman_method". A list of authors is available in Wikipedia. |