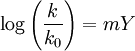To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
SN1 reactionThe SN1 reaction is a substitution reaction in organic chemistry. "SN" stands for nucleophilic substitution and the "1" represents the fact that the rate-determining step is unimolecular [1] [2]. It involves a carbocation intermediate and is commonly seen in reactions of secondary or tertiary alkyl halides or, under strongly acidic conditions, with secondary or tertiary alcohols. With primary alkyl halides, the alternative SN2 reaction occurs. Among inorganic chemists, SN1 is referred to perhaps more accessibly as a dissociative mechanism. A reaction mechanism was first proposed by Christopher Ingold et al in 1940 [3]
Product highlight
MechanismThe SN1 reaction between a molecule A and a nucleophile B takes place in three steps:
KineticsIn contrast to SN2, SN1 reactions take place in two steps (excluding any protonation or deprotonation). The rate determining step is the first step, so the rate of the overall reaction is essentially equal to that of carbocation formation and does not involve the attacking nucleophile. Thus nucleophilicity is irrelevant and the overall reaction rate depends on the concentration of the reactant only.
In 1954 it was found that addition of a small amount of lithium perchlorate certain acetolysis reactions (for example that of the tosylate of cholesterol) led to a remarkable reaction rate increase [5]. Based on this special salt effect the general mechanism was refined to include a contact ion pair (CIP) with cation and anion together in a solvent cage which then dissociates to a so-called solvent-separated ion pair (SSIP) and then on to free ions (FI). All the interconversions are reversible and the added salt prevents the reformation of CIP from SSIP. In some cases the SN1 reaction will occur at an abnormally high rate due to neighbouring group participation (NGP). NGP often lowers the energy barrier required for the formation of the carbocation intermediate. Scope of the reactionThe SN1 mechanism tends to dominate when the central carbon atom is surrounded by bulky groups because such groups sterically hinder the SN2 reaction. Additionally, bulky substituents on the central carbon increase the rate of carbocation formation because of the relief of steric strain that occurs. The resultant carbocation is also stabilized by both inductive stabilization and hyperconjugation from attached alkyl groups. The Hammond-Leffler postulate suggests that this too will increase the rate of carbocation formation. The SN1 mechanism therefore dominates in reactions at tertiary alkyl centers and is further observed at secondary alkyl centers in the presence of weak nucleophiles. StereochemistryBecause the intermediate carbocation is planar, the central carbon is not a stereocenter. Even if it were a stereocenter prior to becoming a carbocation, the original configuration at that atom is lost. Rather, the central carbon can be prochiral. Nucleophilic attack can occur from either side of the plane, so the product might consist of a mixture of two stereoisomers. In fact, if the central carbon is the only stereocenter in the reaction, racemization may occur. This stands in contrast to the SN2 mechanism, where the chiral configuration of the substrate is inverted. However, an excess of inversion is usually observed, as the leaving group can remain in proximity to the carbocation intermediate for a short time and block nucleophilic attack. For example, in the reaction of 3S-chloro-3-methylhexane with iodide ion, if the carbocation intermediate is free of the leaving group then it is achiral and stands an equal chance of attack on either side. This leads to a mixture of 3R-iodo-3-methylhexane and 3S-iodo-3-methylhexane: Side reactionsTwo common side reactions are elimination reactions and carbocation rearrangement. If the reaction is performed under warm or hot conditions (which favor an increase in entropy), E1 elimination is likely to predominate, leading to formation of an alkene. Even if the reaction is performed cold, some alkene may be formed. If an attempt is made to perform an SN1 reaction using a strongly basic nucleophile such as hydroxide or methoxide ion, the alkene will again be formed, this time via an E2 elimination. This will be especially true if the reaction is heated. Finally, if the carbocation intermediate can rearrange to a more stable carbocation, it will give a product derived from the more stable carbocation rather than the simple substitution product. Solvent effectsSince the SN1 reaction involves formation of an unstable carbocation intermediate in the rate-determining step, anything that can facilitate this will speed up the reaction. The normal solvents of choice are both polar (to stabilise ionic intermediates in general) and protic (to solvate the leaving group in particular). Typical polar protic solvents include water and alcohols, which will also act as nucleophiles. The Y scale correlates solvolysis reaction rates of any solvent (k) with that of a standard solvent (80% v/v ethanol/water) (k0) through
with m a reactant constant (m = 1 for tert-butyl chloride) and Y a solvent parameter [6]For example 100% ethanol gives Y = - 2.3, 50% ethanol in water Y = +1.65 and 15% concentration Y = +3.2 [7] See also
References
Categories: Substitution reactions | Reaction mechanisms |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "SN1_reaction". A list of authors is available in Wikipedia. |