To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Calcium carbonate
Calcium carbonate is a chemical compound, with the chemical formula CaCO3. It is a common substance found as rock in all parts of the world, and is the main component of shells of marine organisms, snails, and eggshells. Calcium carbonate is the active ingredient in agricultural lime, and is usually the principal cause of hard water. It is commonly used medicinally as a calcium supplement or as an antacid. Product highlight
OccurrenceCalcium carbonate is found naturally as the following minerals and rocks: To test whether a mineral or rock contains calcium carbonate, strong acids, such as hydrochloric acid, can be added to it. If the sample does contain calcium carbonate, it will fizz and produce carbon dioxide and water. Weak acids such as acetic acid will react, albeit less vigorously. All of the rocks/minerals mentioned above will react with acid. Chemical properties
Calcium carbonate shares the typical properties of other carbonates. Notably:
Calcium carbonate will react with water that is saturated with carbon dioxide to form the soluble calcium bicarbonate.
This reaction is important in the erosion of carbonate rocks, forming caverns, and leads to hard water in many regions. PreparationThe vast majority of calcium carbonate used in industry is extracted by mining or quarrying. Pure calcium carbonate (e.g. for food or pharmaceutical use), can be produced from a pure quarried source (usually marble). Alternatively, calcium oxide is prepared by calcining crude calcium carbonate. Water is added to give calcium hydroxide, and carbon dioxide is passed through this solution to precipitate the desired calcium carbonate, referred to in the industry as precipitated calcium carbonate (PCC):[1]
UsesIndustrial applicationsThe main use of calcium carbonate is in the construction industry, either as a building material in its own right (e.g. marble) or limestone aggregate for roadbuilding or as an ingredient of cement or as the starting material for the preparation of builder's lime by burning in a kiln. Calcium carbonate is also used in the purification of iron from iron ore in a blast furnace. Calcium carbonate is calcined in situ to give calcium oxide, which forms a slag with various impurities present, and separates from the purified iron.[2] Calcium carbonate is widely used as an extender in paints,[3] in particular matte emulsion paint where typically 30% by weight of the paint is either chalk or marble. Calcium carbonate is also widely used as a filler in plastics.[3] Some typical examples include around 15 to 20% loading of chalk in uPVC drain pipe, 5 to 15% loading of stearate coated chalk or marble in uPVC window profile. Fine ground calcium carbonate is an essential ingredient in the microporous film used in babies' diapers and some building films as the pores are nucleated around the calcium carbonate particles during the manufacture of the film by biaxial stretching. Calcium carbonate is also used in a wide range of trade and DIY adhesives, sealants, and decorating fillers.[3] Ceramic tile adhesives typically contain 70 to 80% limestone. Decorating crack fillers contain similar levels of marble or dolomite. It is also mixed with putty in setting stained glass windows, and as a resist to prevent glass from sticking to kiln shelves when firing glazes and paints at high temperature. Calcium carbonate is known as whiting in ceramics/glazing applications,[3] where it is used as a common ingredient for many glazes in its white powdered form. When a glaze containing this material is fired in a kiln, the whiting acts as a flux material in the glaze. In North America, calcium carbonate has begun to replace kaolin in the production of glossy paper. Europe has been practicing this as alkaline papermaking or acid-free papermaking for some decades. Carbonates are available in forms: ground calcium carbonate (GCC) or precipitated calcium carbonate (PCC). The latter has a very fine and controlled particle size, on the order of 2 micrometres in diameter, useful in coatings for paper. Used in swimming pools as a pH corrector for maintaining alkalinity "buffer" to offset the acidic properties of the disinfectant agent. It is commonly called chalk as it has been a major component of blackboard chalk. Chalk may consist of either calcium carbonate or gypsum, hydrated calcium sulfate CaSO4·2H2O. Health and dietary applicationsCalcium carbonate is widely used medicinally as an inexpensive dietary calcium supplement or antacid.[4] It may be used as a phosphate binder for the treatment of hyperphosphatemia (primarily in patients with chronic renal failure) when lanthanum carbonate is not prescribed. It is also used in the pharmaceutical industry as an inert filler for tablets and other pharmaceuticals.[5] As a food additive, it is used in some soy milk products as a source of dietary calcium; one study concludes that calcium carbonate is as bioavailable as ordinary cow's milk.[6] Ecological applicationsIn 1989, a researcher, Ken Simmons, introduced CaCO3 into the Whetstone Brook in Massachusetts.[7] His hope was that the calcium carbonate would counter the acid in the stream from acid rain and save the trout that had ceased to spawn. Although his experiment was a success, it did increase the amounts of aluminium ions in the area of the brook that was not treated with the limestone. This shows that CaCO3 can be added to neutralize the effects of acid rain in river ecosystems. Currently calcium carbonate is used to neutralize acidic conditions in both soil and water.[8][9] Calcination equilibrium
Calcination of limestone using charcoal fires to produce quicklime has been practiced since antiquity by cultures all over the world. The temperature at which limestone yields calcium oxide is usually given as 825 °C, but stating an absolute threshold is misleading. Calcium carbonate exists in equilibrium with calcium oxide and carbon dioxide at any temperature. At each temperature there is a partial pressure of carbon dioxide that is in equilibrium with calcium carbonate. At room temperature the equilibrium overwhelmingly favors calcium carbonate, because the equilibrium CO2 pressure is only a tiny fraction of the partial CO2 pressure in air, which is about 0.035 kPa. At temperatures above 550 °C the equilibrium CO2 pressure begins to exceed the CO2 pressure in air. So above 550 °C, calcium carbonate begins to outgas CO2 into air. But in a charcoal fired kiln, the concentration of CO2 will be much higher than it is in air. Indeed if all the oxygen in the kiln is consumed in the fire, then the partial pressure of CO2 in the kiln can be as high as 20 kPa. The table shows that this equilibrium pressure is not achieved until the temperature is nearly 800 °C. For the outgassing of CO2 from calcium carbonate to happen at an economically useful rate, the equilibrium pressure must significantly exceed the ambient pressure of CO2. And for it to happen rapidly, the equilibrium pressure must exceed total atmospheric pressure of 101 kPa, which happens at 898 °C. SolubilityWith varying CO2 pressure
Calcium carbonate is poorly soluble in pure water. The equilibrium of its solution is given by the equation (with dissolved calcium carbonate on the right):
where the solubility product for [Ca2+][CO32–] is given as anywhere from Ksp = 3.7×10–9 to Ksp = 8.7×10–9 at 25 °C, depending upon the data source.[11][10] What the equation means is that the product of molar concentration of calcium ions (moles of dissolved Ca2+ per liter of solution) with the molar concentration of dissolved CO32– cannot exceed the value of Ksp. This seemingly simple solubility equation, however, must be taken along with the more complicated equilibrium of carbon dioxide with water (see carbonic acid). Some of the CO32– combines with H+ in the solution according to:
HCO3– is known as the bicarbonate ion. Calcium bicarbonate is many times more soluble in water than calcium carbonate -- indeed it exists only in solution. Some of the HCO3– combines with H+ in solution according to:
Some of the H2CO3 breaks up into water and dissolved carbon dioxide according to:
And dissolved carbon dioxide is in equilibrium with atmospheric carbon dioxide according to:
For ambient air,
(which is true for all aqueous solutions), and the fact that the solution must be electrically neutral,
make it possible to solve simultaneously for the remaining five unknown concentrations (note that the above form of the neutrality equation is valid only if calcium carbonate has been put in contact with pure water or with a neutral pH solution; in the case where the origin water solvent pH is not neutral, the equation is modified). The table on the right shows the result for [Ca2+] and [H+] (in the form of pH) as a function of ambient partial pressure of CO2 (Ksp = 4.47×10−9 has been taken for the calculation). At atmospheric levels of ambient CO2 the table indicates the solution will be slightly alkaline. The trends the table shows are
The effect of the latter is especially evident in day to day life of people who have hard water. Water in aquifers underground can be exposed to levels of CO2 much higher than atmospheric. As such water percolates through calcium carbonate rock, the CaCO3 dissolves according to the second trend. When that same water then emerges from the tap, in time it comes into equilibrium with CO2 levels in the air by outgassing its excess CO2. The calcium carbonate becomes less soluble as a result and the excess precipitates as lime scale. This same process is responsible for the formation of stalactites and stalagmites in limestone caves. Two hydrated phases of calcium carbonate, monohydrocalcite, CaCO3·H2O and ikaite, CaCO3·6H2O]], may precipitate from water at ambient conditions and persist as metastable phases. With varying pHWe now consider the problem of the maximum solubility of calcium carbonate in normal atmospheric conditions ( showing a quadratic dependence in [H+]. The numerical application with the above values of the constants gives
Comments:
Solubility in a strong or weak acid solutionSolutions of strong (HCl) or weak (acetic, phosphoric) acids are commercially available. They are commonly used to remove limescale deposits. The maximum amount of CaCO3 that can be "dissolved" by one liter of an acid solution can be calculated using the above equilibrium equations.
where the initial state is the acid solution with no Ca2+ (not taking into account possible CO2 dissolution) and the final state is the solution with saturated Ca2+. For strong acid concentrations, all species have a negligible concentration in the final state with respect to Ca2+ and A− so that the neutrality equation reduces approximately to 2[Ca2+] = [A−] yielding
We see that for the same total acid concentration, the initial pH of the weak acid is less acid than the one of the strong acid; however, the maximum amount of CaCO3 which can be dissolved is approximately the same. This is because in the final state, the pH is larger that the pKA, so that the weak acid is almost completely dissociated, yielding in the end as many H+ ions as the strong acid to "dissolve" the calcium carbonate.
where [A] = [H3PO4] + [H2PO4−] + [HPO42−] + [PO43−]. We see that phosphoric acid is more efficient than a monoacid since at the final almost neutral pH, the second dissociated state concentration [HPO42−] is not negligible (see phosphoric acid ). See also
References
Categories: Calcium compounds | Carbonates | Limestone | Phosphate binders |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Calcium_carbonate". A list of authors is available in Wikipedia. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



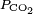 (atm)
(atm)
![\frac{P_{\mathrm{CO}_2}}{[\mathrm{CO}_2]}\ =\ k_\mathrm{H}](images/math/3/b/b/3bb040ed9d2fa386e59989866610188f.png)
![[\mathrm{Ca}^{2+}]_\mathrm{max} = \frac{K_\mathrm{sp}k_\mathrm{H}} {K_\mathrm{h}K_\mathrm{a1}K_\mathrm{a2}} \frac{[\mathrm{H}^+]^2}{P_{\mathrm{CO}_2}}](images/math/5/0/4/504308c38c93c64347f29e110b98498c.png)
![\scriptstyle[\mathrm{Ca}^{2+}] \simeq \frac{[\mathrm{A}^-]}{2}](images/math/b/e/2/be27098c6038ef4b45348e8acc57825e.png) . When the concentration decreases, [HCO3−] becomes non negligible so that the preceding expression is no longer valid. For vanishing acid concentrations, we recover the final pH and the solubility of CaCO3 in pure water.
. When the concentration decreases, [HCO3−] becomes non negligible so that the preceding expression is no longer valid. For vanishing acid concentrations, we recover the final pH and the solubility of CaCO3 in pure water.
Last viewed


