Chemical reaction design goes virtual
Researchers aim to streamline the time- and resource-intensive process of screening ligands during catalyst design by using virtual ligands
Advertisement



Researchers at the Institute for Chemical Reaction Design and Discovery and Hokkaido University have developed a virtual ligand-assisted (VLA) screening method, which could drastically reduce the amount of trial and error required in the lab during transition metal catalyst development. The method, published in the journal ACS Catalysis, may also lead to the discovery of unconventional catalyst designs outside the scope of chemists’ intuition.
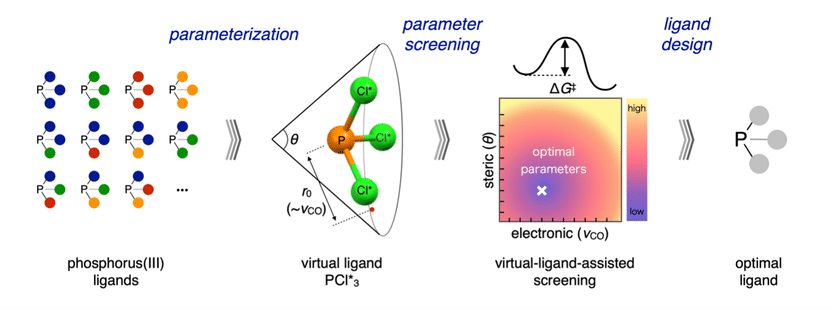
VLA screening workflow. A ligand class (left) is parameterized into a virtual ligand (middle left). Parameters are screened to produce a contour map (middle right), which is used to rationally design an optimal ligand (right).
Wataru Matsuoka, Yu Harabuchi, Satoshi Maeda. ACS Catalysis. March 13, 2022
Ligands are molecules that are bonded to the central metal atom of a catalyst, and they affect the activity and selectivity of a catalyst. Finding the optimal ligand to catalyze a specific target reaction can be like finding a needle in a haystack. The VLA screening method provides a way to efficiently search that haystack, surveying a broad range of values for different properties to identify the features of ligands that should be most promising. This narrows down the search area for chemists in the lab and has the potential to greatly accelerate the reaction design process.
This new work utilizes virtual ligands, which mimic the presence of real ligands; however, instead of being described by many individual constituent atoms—such as carbon or nitrogen—virtual ligands are described using only two metrics: their steric, or space-filling, properties and their electronic properties. Researchers developed approximations that describe each of these effects with a single parameter. Using this simplified description of a ligand enabled researchers to evaluate ligands in a computationally efficient way over a large range of values for these two effects. The result is a “contour map” that shows what combination of steric and electronic effects a ligand should have in order to best catalyze a specific reaction. Chemists can then focus on only testing real ligands that fit these criteria.

Researchers used monodentate phosphorus (III) virtual ligands as a test group and verified their models for the electronic and steric properties of the virtual ligands against values calculated for corresponding real ligands.
The VLA screening method was then employed to design ligands for a test reaction in which a CHO group and a hydrogen atom can be added to a double bond in two different possible configurations. The reaction pathway was evaluated for 20 virtual ligand cases (consisting of different assigned values for the electronic and steric parameters) to create a contour map that shows a visual trend for what types of ligands can be expected to result in a highly selective reaction.
Computer models of real ligands were designed based on parameters extracted from the contour map and then evaluated computationally. The selectivity values predicted via the VLA screening method matched well with the values computed for the models of real ligands, showing the viability of the VLA screening method to provide guidance that aids in rational ligand design.
Beyond saving valuable time and resources, corresponding author Satoshi Maeda anticipates the creation of powerful reaction prediction systems by combining the VLA screening method with other computational techniques.
“Ligand screening is a pivotal process in the development of transition metal catalysis. As the VLA screening can be conducted in silico, it would save a lot of time and resources in the lab. We believe that this method not only streamlines finding an optimal ligand from a given library of ligands, but also stimulates researchers to explore the untapped chemical space of ligands,” commented corresponding author Satoshi Maeda. “Furthermore, we also expect that by combining this method with our reaction prediction technology using the Artificial Force Induced Reaction method, a new computer-driven discovery scheme of transition metal catalysis can be realized.”
Original publication



Other news from the department science

























































