When changing one atom makes molecules better
A method to replace hydrogen with fluorine in organic molecules makes drugs more effective
Advertisement



The development and improvement of pharmaceuticals plays the central role in the ongoing battle against human disease. organic synthesis is the field that enables these developments as it offers the toolbox to diversify chemical structures. The group of Nuno Maulide, recently named the Scientist of the Year 2018 in Austria, in collaboration with the group of Harald Sitte, has now reported a facile method for the replacement of hydrogen with fluorine in important drug molecules. This new discovery enables the fine-tuning of existing (and potential new) pharmaceuticals to endow them with improved pharmacological properties.
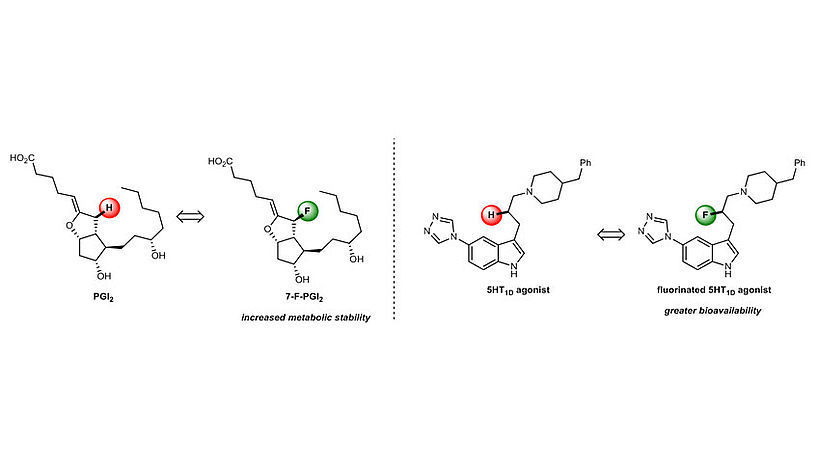
© Universität Wien/Maulide Group
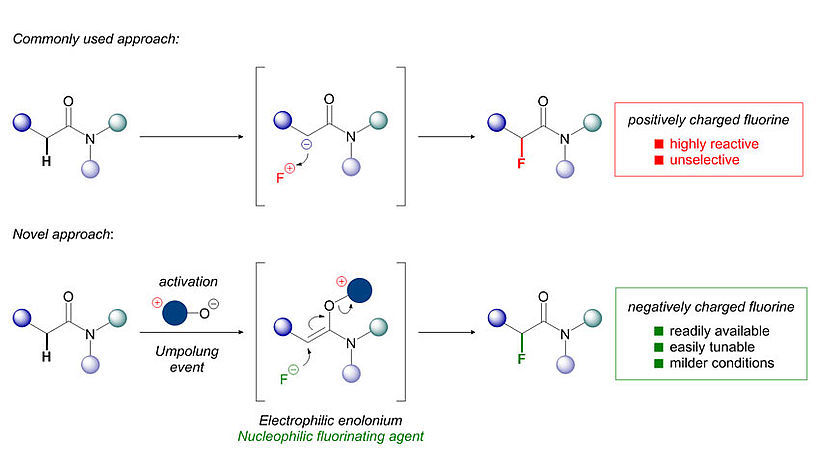
© Universität Wien/Maulide Group
The vast majority of pharmaceuticals employed in the treatment of human diseases are of an organic nature, meaning that the active component is a molecule (or a combination of several molecules) that is constituted of carbon and hydrogen atoms. This trait is shared with all living matter, as proteins, sugars, fats and DNA are also based on such hydrocarbon skeletons, differing only by arrangement and their substitution with a relatively small amount of other possible elements (mainly oxygen, nitrogen, sulfur and phosphorus). "Our bodies are nothing else than a large assembly of billions of carbon-made, or in other words, organic molecules", says Nuno Maulide, recently named Scientist of the Year 2018 in Austria and Professor at the University of Vienna. Owing to this similarity, organic pharmaceuticals are ideally suited to interact with the human body, for example by binding to receptors thereby triggering or inhibiting a desired or undesired function.
Finding the key that best fits the lock
The design of a pharmaceutical molecule targeting a specific interaction with a receptive structure is often conceptualised by the analogy of a lock and key. "The receptor (for example an enzyme) has a unique structure (lock) and therefore requires a unique structure (key) to interact with. Due to the need for an exact fit, the structural integrity of the pharmaceutical compound is key (pun intended!) to assure its beneficial bioactivity", explains Harald Sitte, Professor at the Medical University of Vienna and co-author of the study.

Trying to escape the clean-up
However, just as nutrients are metabolised by the body, pharmaceuticals that enter the body (made up of the same essential constituents, carbon, hydrogen, …) are also degraded by the same enzymes that metabolise and eliminate our food constituents. “This type of clean-up machinery is essential for our body in order to protect itself; molecules that are undesired and could have negative effects must be eliminated swiftly. Unfortunately, such machinery is often indiscriminate and pharmaceuticals will also be metabolised as soon as they enter in contact with the body”, explains Christopher Teskey, PostDoc in the Maulide group and joint first author of the study. This can alter the structure of pharmaceuticals and therefore also remove their beneficial properties. "Large parts of this degradation occur precisely at the junction of carbon and hydrogen atoms (C–H bonds), that can be broken or altered to form new compounds that can be more easily cleared from the body by excretion. C–H bonds are inherently rather weak bonds, meaning that oxidation can take place easily", explains Pauline Adler, former PostDoc in the Maulide group and the study’s other joint first author. "It ultimately is a race to escape cleaning up! The longer a beneficial pharmaceutical can escape this cat-and-mouse chase with the metabolism enzymes, the longer its beneficial effect can be felt in the body", jokes Maulide.
A clever solution: exchange H with F can be a 2-in-1
It is therefore obvious that, if the structural weak points of drug molecules could be eliminated or mitigated, their metabolic stability could be significantly increased. Chemists have found some years ago that the strategic replacement of especially weak C–H bonds with much stronger C–F bonds can be a highly rewarding approach in this direction. While hydrogen and fluorine are distinctly different in some respects, their sizes are comparable, and the replacement of H with F can therefore often be assumed to have only minimal effect on the structure of the key (pharmaceutical). "Even more so, because of these differed electronic properties, a strategically placed fluorine atom can potentially establish additional interactions with its target (lock), thereby enhancing the desired activity", explains Maulide. "Moreover, introduction of fluorine into a drug molecule can alter its properties in such a way that it can be more easily absorbed by the body (increased bioavailablility), once again increasing the amount of drug that can find and interact with its receptor. It really can be a 2-in-1 situation!", posits Daniel Kaiser, former PhD student of the Maulide group and co-author of the study.
The challenge of replacing H with F: if only you could use toothpaste
While the introduction of fluorine can have this wide range of beneficial effects on a pharmaceutical, its placement onto organic molecules is often far from trivial. The most common methods of fluorination involve highly reactive, corrosive and sometimes toxic reagents. These reagents are based on positively charged fluorine atoms (F+), which are far more expensive and difficult to handle than their common and cheap negatively charged (F–) fluoride counterparts (the fluoride anion itself is commonly known, for example, as a constituent of toothpaste).
A simple solution and the first demonstration
The research group of Nuno Maulide (Institute of Organic Chemistry, University of Vienna) has now discovered a facile and selective method for the introduction of fluorine atoms into organic molecules using the widely available fluoride anion (F–). "Most chemists have looked to introduce fluorine by using negatively polarised organic molecules that can react with a F+ reagent. We simply did the opposite: switch polarity of the organic molecule so that we can use the same fluoride you have in toothpaste!", enthuses Maulide. Importantly, this approach employs cheap starting materials, is operationally simple and delivers high yields of the products in short amounts of time.
In the published work, several fluorinated analogues of common bioactive agents were easily synthesised, most prominently fluoro-Citalopram. "The parent molecule Citalopram is a blockbuster antidepressant used in the treatment of clinical depression. It interacts with the serotonin transporter (SERT) and raises the synaptic concentration of serotonin to alleviate the depressive symptoms", explains Sitte. In collaboration with the group of Prof. Harald Sitte (MedUni Vienna), Prof. Maulide and his team were able to establish that the activity of Citalopram is retained upon insertion of a fluorine atom. Significantly, while the activity is retained (despite the structural change), other pharmacological factors, such as metabolic stability and bioavailability, are expected to improve as a consequence of fluorination. The teams therefore believe that fluoro-Citalopram could present a viable alternative to the non-fluorinated analogue.
Chemistry opens the door
"Having a method to exchange H with F under such simple conditions is but the beginning. We can now imagine doing this exchange in a range of other pharmaceuticals and study the properties of the resulting, new analogues. Since the exchange of H with F is also a technique that can be of relevance to the materials industry, you can understand why we are excited about this work", explains Maulide. "This is a great example of the power of Chemistry: because we are able to manipulate the structure of matter at a molecular level with atomic precision, we can open doors that would otherwise remain closed to the imagination", closes Maulide.
Original publication



Other news from the department science





















Most read news









More news from our other portals





























See the theme worlds for related content
Topic world Synthesis
Chemical synthesis is at the heart of modern chemistry and enables the targeted production of molecules with specific properties. By combining starting materials in defined reaction conditions, chemists can create a wide range of compounds, from simple molecules to complex active ingredients.

Topic world Synthesis
Chemical synthesis is at the heart of modern chemistry and enables the targeted production of molecules with specific properties. By combining starting materials in defined reaction conditions, chemists can create a wide range of compounds, from simple molecules to complex active ingredients.