Machine learning technique speeds up crystal structure determination
Advertisement


Nanoengineers at the University of California San Diego have developed a computer-based method that could make it less labor-intensive to determine the crystal structures of various materials and molecules, including alloys, proteins and pharmaceuticals. The method uses a machine learning algorithm, similar to the type used in facial recognition and self-driving cars, to independently analyze electron diffraction patterns, and do so with at least 95% accuracy.
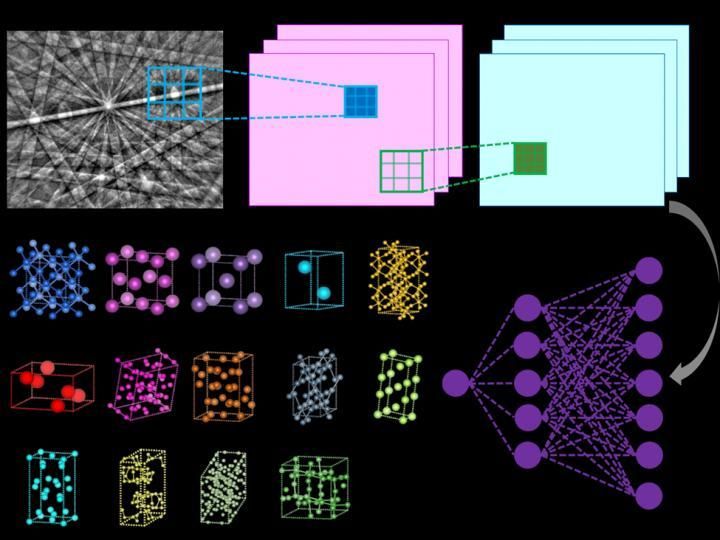
Illustration of the inner workings of a convolutional neural network that computes the probability that the input diffraction pattern belongs to a given class (e.g. Bravais lattice or space group).
Vecchio lab/Science
A team led by UC San Diego nanoengineering professor Kenneth Vecchio and his Ph.D. student Kevin Kaufmann, who is the first author of the paper, developed the new approach. Their method involves using a scanning electron microscope (SEM) to collect electron backscatter diffraction (EBSD) patterns. Compared to other electron diffraction techniques, such as those in transmission electron microscopy (TEM), SEM-based EBSD can be performed on large samples and analyzed at multiple length scales. This provides local sub-micron information mapped to centimeter scales. For example, a modern EBSD system enables determination of fine-scale grain structures, crystal orientations, relative residual stress or strain, and other information in a single scan of the sample.
However, the drawback of commercial EBSD systems is the software's inability to determine the atomic structure of the crystalline lattices present within the material being analyzed. This means a user of the commercial software must select up to five crystal structures presumed to be in the sample and then the software attempts to find probable matches to the diffraction pattern. The complex nature of the diffraction pattern often causes the software to find false structure matches in the user selected list. As a result, the accuracy of the existing software's determination of the lattice type is dependent on the operator's experience and prior knowledge of their sample.
The method that Vecchio's team developed does this all autonomously, as the deep neural network independently analyzes each diffraction pattern to determine the crystal lattice, out of all possible lattice structure types, with a high degree of accuracy (greater than 95%).
A wide range of research areas including pharmacology, structural biology, and geology are expected to benefit from using similar automated algorithms to reduce the amount of time required for crystal structural identification, researchers said.
Original publication
"Crystal symmetry determination in electron diffraction using machine learning"; Science; 2020

Other news from the department science