Highly Accurate Results at Low Computational Cost
Researchers Succeed in Predicting NMR Parameters with DLPNO Approximation
Advertisement



Nuclear magnetic resonance (NMR) is one of the most important analytical techniques used in chemical, pharmaceutical, and biomedical research as well as in materials sciences. It provides detailed information about the structures of molecules in solution, which is, however, indirect. Hence, researchers often have to rely on tools from theoretical chemistry in order to properly interpret the complex experimental data. One of the primary sources of information in NMR spectra is the so-called “chemical shift” that exists for each atom in a molecule and depends on its surroundings. Quantum chemists simulate the chemical shift for each atom, in order to receive the full information content of NMR spectra. However, the methods used so far repeatedly reached their limits.
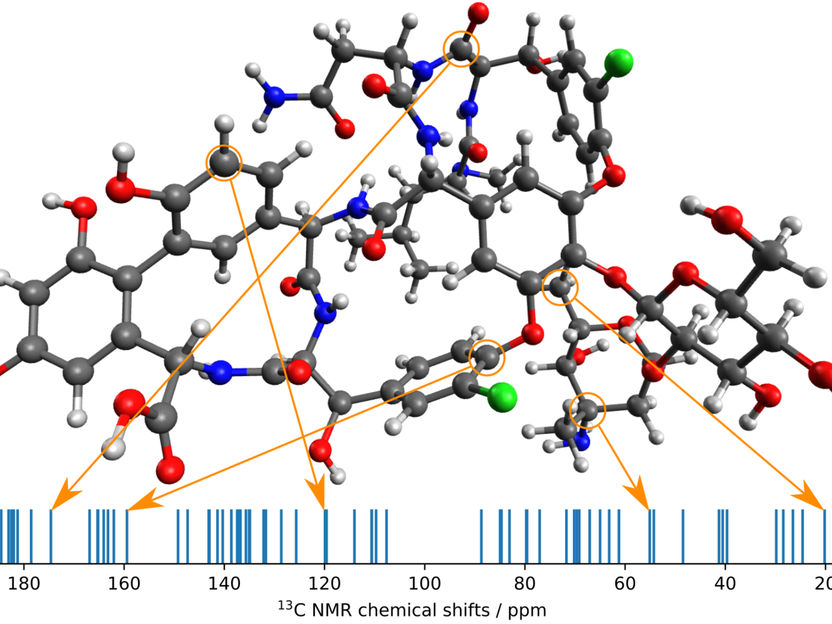
The MPI researchers’ new method in application. A conformer of the vancomycin molecule and its computed 13C NMR spectrum with some peaks assigned to atoms.
© MPI für Kohlenforschung
Previous results were often too inaccurate or needed high computational effort - the new DLPNO approximation significantly reduces the computational effort of exact methods
The combination of experiment and new theoretical methods leads to insights that would not have been accessible by other means. However, scientists often face a dilemma: efficient methods such as density functional theory - the workhorse of theoretical chemistry – regularly provide insufficient accuracy. High-accuracy wavefunction methods, in turn, require far more computational resources and their scope is restricted to molecules with only a few atoms – far too small for successful applications in pharmacy or biomedical research.
Researchers from the Department of Molecular Theory and Spectroscopy at the MPI für Kohlenforschung have now developed a new approach that brings a breakthrough. Their wave function method is based on the so-called domain based local pair natural orbital (DLPNO) framework, a concept developed by Frank Neese. In a recently published article, Stoychev et al. report how they succeeded in accurately calculating chemical shifts for systems with hundreds of atoms. Their new method saves computational time and opens up interesting perspectives for users, because in many areas of chemistry, the interpretation of NMR spectra is part of daily business.
Original publication



Other news from the department science





















Most read news











More news from our other portals




























See the theme worlds for related content
Topic World Spectroscopy
Investigation with spectroscopy gives us unique insights into the composition and structure of materials. From UV-Vis spectroscopy to infrared and Raman spectroscopy to fluorescence and atomic absorption spectroscopy, spectroscopy offers us a wide range of analytical techniques to precisely characterize substances. Immerse yourself in the fascinating world of spectroscopy!

Topic World Spectroscopy
Investigation with spectroscopy gives us unique insights into the composition and structure of materials. From UV-Vis spectroscopy to infrared and Raman spectroscopy to fluorescence and atomic absorption spectroscopy, spectroscopy offers us a wide range of analytical techniques to precisely characterize substances. Immerse yourself in the fascinating world of spectroscopy!