Calculating the "fingerprints" of molecules with artificial intelligence
Advertisement



With conventional methods, it is extremely time-consuming to calculate the spectral fingerprint of larger molecules. But this is a prerequisite for correctly interpreting experimentally obtained data. Now, a team at HZB has achieved very good results in significantly less time using self-learning graphical neural networks.
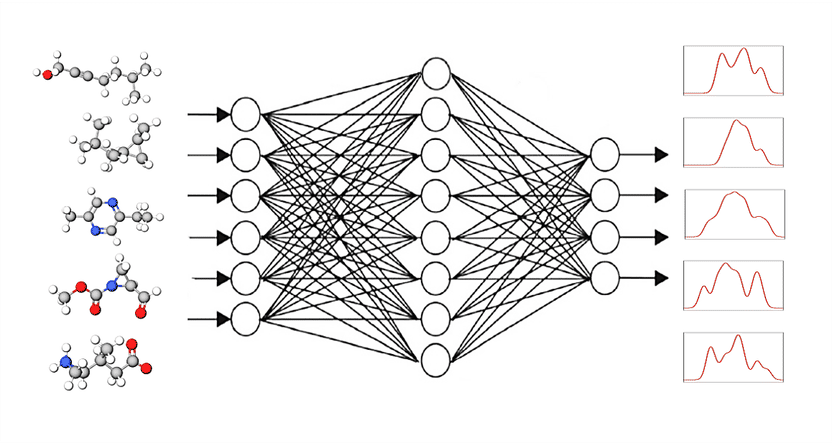
The graphical neural network GNN receives small molecules as input with the task of determining their spectral responses. By matching them with the known spectra, the GNN programme learns to calculate spectra reliably.
© K. Singh, A. Bande/HZB
"Macromolecules but also quantum dots, which often consist of thousands of atoms, can hardly be calculated in advance using conventional methods such as DFT," says PD Dr. Annika Bande at HZB. With her team she has now investigated how the computing time can be shortened by using methods from artificial intelligence.
The idea: a computer programme from the group of "graphical neural networks" or GNN receives small molecules as input with the task of determining their spectral responses. In the next step, the GNN programme compares the calculated spectra with the known target spectra (DFT or experimental) and corrects the calculation path accordingly. Round after round, the result becomes better. The GNN programme thus learns on its own how to calculate spectra reliably with the help of known spectra.
"We have trained five newer GNNs and found that enormous improvements can be achieved with one of them, the SchNet model: The accuracy increases by 20% and this is done in a fraction of the computation time," says first author Kanishka Singh. Singh participates in the HEIBRiDS graduate school and is supervised by two experts from different backgrounds: computer science expert Prof. Ulf Leser from Humboldt University Berlin and theoretical chemist Annika Bande.
"Recently developed GNN frameworks could do even better," she says. "And the demand is very high. We therefore want to strengthen this line of research and are planning to create a new postdoctoral position for it from summer onwards as part of the Helmholtz project "eXplainable Artificial Intelligence for X-ray Absorption Spectroscopy"."
Original publication




Other news from the department science





















Most read news












More news from our other portals



























See the theme worlds for related content
Topic World Spectroscopy
Investigation with spectroscopy gives us unique insights into the composition and structure of materials. From UV-Vis spectroscopy to infrared and Raman spectroscopy to fluorescence and atomic absorption spectroscopy, spectroscopy offers us a wide range of analytical techniques to precisely characterize substances. Immerse yourself in the fascinating world of spectroscopy!

Topic World Spectroscopy
Investigation with spectroscopy gives us unique insights into the composition and structure of materials. From UV-Vis spectroscopy to infrared and Raman spectroscopy to fluorescence and atomic absorption spectroscopy, spectroscopy offers us a wide range of analytical techniques to precisely characterize substances. Immerse yourself in the fascinating world of spectroscopy!
