Memory matters for quantum atomic motion on metals
New findings with implications for heterogeneous catalysis and energy storage
Advertisement



George Trenins and Mariana Rossi of the Lise Meitner Group at the Max Planck Institute for the Structure and Dynamics of Matter (MPSD) have developed an efficient technique for simulating the impact of electronic friction on quantum atomic motion at metallic interfaces. Applying it to hydrogen atoms on copper, they explain the physical origin of experimentally observed diffusion rates — offering new insight with implications for heterogeneous catalysis and energy storage. Their work has now been published in Physical Review Letters (PRL).
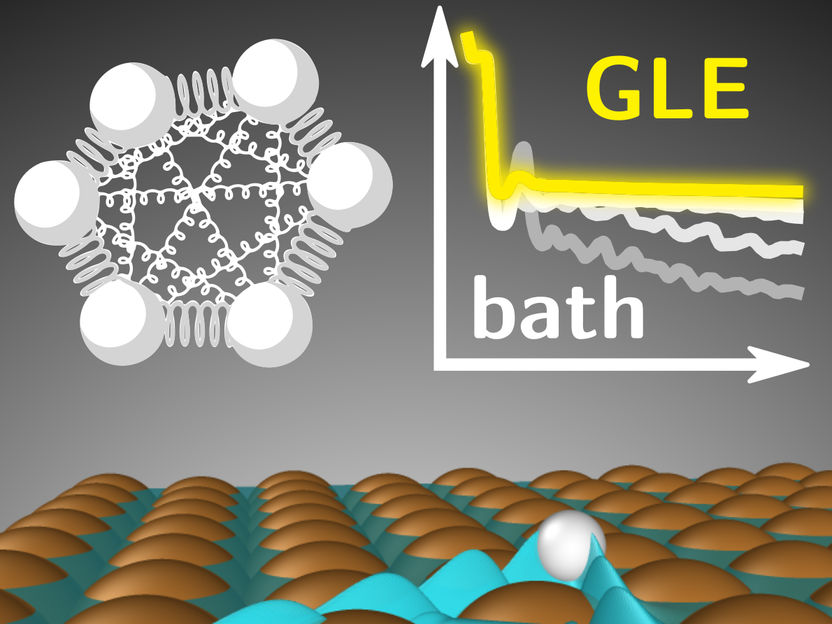
Electronic friction is the main energy dissipation mechanism for hydrogen atoms diffusing on copper surfaces. Within path-integral theory, its influence on nuclear quantum effects is simulated with "ring polymers" (left) moving according to the generalized Langevin equation (GLE). The new work enables such dynamics simulations for complex molecular systems.
G. Trenins
In a variety of technological applications related to chemical energy generation and storage, atoms and molecules diffuse and react on metallic surfaces. Being able to simulate and predict this motion is crucial to understanding material degradation, chemical selectivity, and to optimizing the conditions of catalytic reactions. Central to this is a correct description of the constituent parts of atoms: electrons and nuclei.
An electron is incredibly light–its mass is almost 2,000 times smaller than that of even the lightest nucleus. This mass disparity allows electrons to adapt rapidly to changes in nuclear positions, which usually enables researchers to use a simplified “adiabatic” description of atomic motion. While this can be an excellent approximation, in some cases the electrons are affected by nuclear motion to such an extent that we need to abandon this simplification and account for the coupling between the dynamics of electrons and nuclei, leading to so-called “non-adiabatic effects.”
One kind of non-adiabatic effect that is especially relevant to metals is termed electronic friction. It affects the probabilities of atoms and molecules sticking to metals, the rate at which they dissipate energy via vibrations, and the speed at which they diffuse on metallic surfaces. If nuclei were classical particles, this phenomenon could be described as the electrons exerting a drag force on the nuclei as they move through a metallic environment.
However, atomic nuclei are quantum objects whose motion does not accord with our everyday intuition and is challenging to simulate on a computer. Unlike classical objects, atomic nuclei possess inherent zero-point energy, making it easier for them to escape a potential-energy well. They can also undergo quantum tunnelling, leaving the well even if they lack sufficient energy to overcome the surrounding barrier. These effects alter chemical reaction rates by orders of magnitude and must be factored into simulations to gain reliable physical insight.
In their recent work, George Trenins and Mariana Rossi succeeded in combining electronic friction with a practical methodology for simulating nuclear quantum effects, based on the path-integral formulation of quantum mechanics. Importantly, their approach can describe frictional forces that depend not only on the current state of the atoms, but also on the trajectory they followed in the past. A combination of this property, known as “memory,” with zero-point energy effects allowed the researchers to explain the unexpected agreement between earlier classical simulations and experimental results.
“Our approach aims to get the right answer for the right reason”, says Trenins. “By capturing the interplay between quantum nuclei and electronic friction, we can gain deep insight into energy exchange at conducting surfaces and aid the design of novel heterogeneous catalysts, such as single-atom alloys and two-dimensional materials.” Mariana Rossi adds, “It is definitely exciting to have a method that can be applied to more complex systems in their full dimensionality. This will allow us to explore these effects in systems that were previously not amenable to such simulations.”
Original publication



Other news from the department science



























































