To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Activity (chemistry)Activity in chemistry is a measure of how different molecules in a non-ideal gas or solution interact with each other. Activity effects are the result of interactions between ions both electrostatic and covalent. The activity of an ion is influenced by its surroundings. The activity of an ion in a cage of water molecules is different from that in the middle of a counterion cloud. This type of activity is relevant throughout chemistry from reaction rates to equilibrium constants. For instance large deviations can exist between the calculated hydrogen ion concentration of a strong acid in solution and the hydrogen activity derived from a pH meter or a pH indicator.[1] Product highlight
DefinitionIn ideal mixtures we can write the dependency of the thermodynamic potential on the composition (written as mole fraction x) as: We can use this formula to define an activity a, by insisting that the same formulism continues to hold in a non-ideal case. Obviously a=x in the ideal case. We can also define an activity coefficient γ:
However there are many alternative schemes to define activity. For dilute solutions usually the solvent follows Raoult's law, but the solute follows Henry's law. It is often convenient to use these laws as a basis for a definition:
Define activities:
As Ksolute ≠ P*solute this implies the choice of a different reference point. It represents the extrapolation of the Henry line all the way to x1 =1 and this is a virtual state: a state of the system that can never be achieved. However, the definition leads to a consistent system of values as long as we do not make x1 too large. The same holds if we use molarities or molalities as concentration measures rather than mole fractions. We can safely use such definitions but the meaning of the plimsoll superscript Molalities are often preferred in this type of calculation because the volumes of non-ideal mixtures are strictly spoken not additive. Molalities do not depend on volume, molarities do. For a gaseous mixtureThe activity is of the form: where γi is the dimensionless fugacity coefficient (fugacity) of the species i, xi the mole fraction of the species in the gaseous mixture and p the pressure expressed in bar. The quantity fi has the dimension of pressure and is fugacity of the gas: for a pure Ideal gas, the fugacity coefficient is equal to one. The value p0 is the standard atmospheric pressure. It is equal to 1 bar and is present to cancel the dimensions of the formula. For compound in a liquid solutionThe activity is of the form: Where γi is the coefficient of activity of the species i, Ci is concentration in one of its many measures. If we (somewhat unwisely) choose to work in molarities the C If more than one concentration is to be taken into account, it is in principle possible to do one in molality, the other in mole fraction etc. This does lead to a legitimate choice of standard state and a consistent set of values. (Obviously one might have to convert if comparing with someone else's numbers that are based on a more obvious choice like everything in molality). In any case, the nature of Ionic solutionsWhen the solute undergoes ionic dissociation in solution (a salt e.g.), the system becomes decidedly non-ideal and we need to take the dissociation process into consideration. We can define activities for the cations and anions separately (a+ and a-). It should be noted however that in a liquid solution the activity coefficient of a given ion (e.g. Ca2+) isn't measurable because it is experimentally impossible to independently measure the electrochemical potential of an ion in solution. (We cannot add cations without putting in anions at the same time). Therefore one introduces the notions of
Even though γ+ and γ- cannot be determined separately, γ± is a measureable quantity that can also be predicted for sufficiently dilute systems using Debye-Hückel theory. For the activity of a strong ionic solute (complete dissociation) we can write:
MeasurementThe most direct way of measuring an activity of a species is to measure its partial vapor pressure in equilibrium with a number of solutions of different strength. For some solutes this is not practical, say sucrose or salt (NaCl) do not have a measurable vapor pressure at ordinary temperatures. However, in such cases it is possible to measure the vapor pressure of the solvent instead. Using the Gibbs-Duhem relation it is possible to translate the change in solvent vapor pressures with concentration into activities for the solute. Another way to determine the activity of a species is through the manipulation of colligative properties, specifically freezing point depression. Using freezing point depression techniques, it is possible to calculate the activity of a weak acid from the relation,
There are also electrochemical methods that allow the determination of activity and its coefficient. The value of the mean ionic activity coefficient γ± of ions in solution can also be estimated with the Debye-Hückel equation, the Davies equation or the Pitzer equation. UseChemical activities should be used to define chemical potentials, where the chemical potential depends on the temperature T, pressure p and the activity ai according to the formula: where R is the Ideal Gas Constant and µi The chemical activities should be used to define the equilibrium constant but in practice often concentrations are used. The same is often true of equtions derived for reaction rates. However, there are circumstances where the activity and the concentration are significantly different and as such, it is not valid to approximate with concentrations where activities are required. Two examples serve this point:
Formulae involving activities can be simplified by considering that:
The latter follows from any definition based on Raoult's law, because if we let the solute concentration x1 go to zero, the vapor pressure of the solvent P will go to P*. Thus its activity a= P/P* will go to unity. This means that if during a reaction in dilute solution more solvent is generated (the reaction produces water e.g.) we can typically set its activity to unity. Solid and liquid activities do not depend very strongly on pressure because their molar volumes are typically small. Graphite at 100 bars has an activity of only 1.01 if we choose 1 bar as standard state. Only at very high pressures do we need to worry about such changes. See alsoReferences
|
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Activity_(chemistry)". A list of authors is available in Wikipedia. |



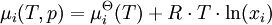
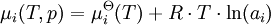
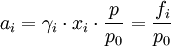
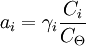
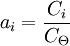
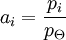
Last viewed


