To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Debye-Hückel equationThe Debye-Hückel limiting law, named for its developers Peter Debye and Erich Hückel, provides one way to obtain activity coefficients . Activities, rather than concentrations, are needed in many chemical calculations because solutions that contain ionic solutes do not behave as ideally even at very low concentrations. The activity is proportional to the concentration by a factor known as the activity coefficient In order to calculate the activity of an ion in a solution, one must know the concentration and the activity coefficient,
The Debye-Hückel limiting law enables one to determine the activity coefficient of an ion in a dilute solution of known ionic strength. The equation is (Hamann, Hamnett, and Vielstich. Electrochemistry. Wiley-VCH. section 2.5.2)
It is important to note that because the ions in the solution act together, the activity coefficient obtained from this equation is actually a mean activity coefficient. Product highlight
Summary of Debye & Hückel's first paper on the theory of dilute electrolytesThe English title of the paper is called "On the Theory of Electrolytes. I. Freezing Point Depression and Related Phenomena." It was originally published in volume 24 of a German-language journal, called Physikalische Zeitschrift, in 1923. An English translation[1]of the paper is included in a book of collected papers presented to Debye by "his pupils, friends, and the publishers on the occasion of his seventieth birthday on March 24, 1954."[1] The paper deals with the calculation of properties of electrolyte solutions that are not under the influence of net electric fields, thus it deals with electrostatics. In the same year they first published this paper, Debye and Hückel, hereinafter D&H, also released a paper that covered their initial characterization of solutions under the influence of electric fields called "On the Theory of Electrolytes. II. Limiting Law for Electric Conductivity," but that subsequent paper is not (yet) covered here. In the following summary (as yet incomplete and unchecked), modern notation and terminology are used, from both chemistry and mathematics, in order to prevent confusion. Also, with a few exceptions to improve clarity, the subsections in this summary are (very) condensed versions of the same subsections of the original paper. IntroductionD&H note that the Guldberg-Waage formula for electrolyte species in chemical reaction equilibrium in classical form is[1]
D&H say that, due to the "mutual electrostatic forces between the ions," it is necessary to modify the Guldberg-Waage equation by replacing K with γK, where γ is an overall activity coefficient, not a "special" activity coefficient (a separate activity coefficient associated for each species)—which is what is used in modern chemistry as of 2007. The relationship between γ and the special activity coefficients, γi is[1]
FundamentalsD&H use the Helmholtz and Gibbs free entropies, Φ and Ξ, to express the effect of electrostatic forces in an electrolyte on its thermodynamic state. Specifically, they split most of the thermodynamic potentials into classical and electrostatic terms.
D&H give the total differential of Φ as[1]
By the definition of the total differential, this means that
which are useful further on. As stated previously, the internal energy is divided into two parts,[1]
Similarly, the Helmholtz free entropy is also divided into two parts,
D&H state, without giving the logic, that[1] It would seem that, without some justification,
Without mentioning it specifically, D&H later give what might be the required (above) justification while arguing that Φe = Ξe. The definition of the Gibbs free entropy, Ξ, is[1]
D&H give the total differential of Ξ as[1]
At this point D&H note that, for water containing 1 mole per liter of potassium chloride (nominal pressure and temperature aren't given), the electric pressure, Pe, amounts to 20 atmospheres. Furthermore, they note that this level of pressure gives a relative volume change of 0.001. Therefore, they neglect change in volume of water due to electric pressure, writing[1]
and put
D&H say that, according to Planck, the classical part of the Gibbs free entropy is[1]
Species zero is the solvent. The definition of ξi is as follows, where lower case letters indicate the particle specific versions of the the corresponding extensive properties:[1]
D&H don't say so, but the functional form for Ξk may be derived from the functional dependence of the chemical potential of a component of an ideal mixture upon its mole fraction.[2] D&H note that the internal energy, U, of a solution is lowered by the electrical interaction of its ions, but that this effect can't be determined by using the crystallographic approximation for distances between dissimilar atoms (the cube root of the ratio of total volume to the number of particles in the volume). This is due to the fact that there is more thermal motion in a liquid solution than in a crystal. The thermal motion tends to smear out the natural lattice that would otherwise be constructed by the ions. Instead, D&H introduce the concept of an ionic cloud. Like the crystal lattice, each ion still attempts to surround itself with oppositely charged ions, but in a more free-form manner; at small distances away from positive ions, one is more likely to find negative ions and vice versa.[1] The Potential Energy of an Arbitrary Ion SolutionElectroneutrality of a solution requires that[1]
To bring an ion of species i, initially far away, to a point P within the ion cloud requires interaction energy in the amount of
at every point in the cloud. Note that in the infinite temperature limit, all ions are distributed uniformly, with no regard for their electrostatic interactions.[1] The charge density is related to the number density:[1]
When combining this result for the charge density with the Poisson equation from electrostatics, a form of the Poisson-Boltzmann equation results:[1]
This equation is difficult to solve and does not follow the principle of linear superposition for the relationship between the number of charges and the strength of the potential field. However, for sufficiently low concentrations of ions, a first order Taylor series approximation for the exponential function may be used (ex = 1 + x for
The Poisson-Boltzmann equation is transformed to[1]
because the first summation is zero due to electroneutrality.[1] Factor out the scalar potential and assign the leftovers, which are constant, to κ2. Also, let I be the ionic strength of the solution:[1]
So, the fundamental equation is reduced to a form of the Helmholtz equation:(http://guava.physics.uiuc.edu/~nigel/courses/569/Essays_2004/files/lu.pdf section 3.1)
Today, κ − 1 is called the Debye screening length. D&H recognize the importance of the parameter in their paper and characterize it as a measure of the thickness of the ion atmosphere, which is (in this paper) an electrical double layer of the Helmholtz type.[1] The equation may be expressed in spherical coordinates by taking r = 0 at some arbitrary ion (http://hyperphysics.phy-astr.gsu.edu/hbase/electric/laplace.html):[1]
The equation has the following general solution; keep in mind that κ is a positive constant:[1]
The electric potential is zero at infinity by definition, so A'' must be zero.[1] In the next step, D&H assume that there is a certain radius, ai, beyond which no ions in the atmosphere may approach the (charge) center of the singled out ion. This radius may be due to the physical size of the ion itself, the sizes of the ions in the cloud, and any water molecules that surround the ions. Mathematically, they treat the singled out ion as a point charge to which one may not approach within the radius ai.[1] The potential of a point charge by itself is:
D&H say that the total potential inside the sphere is[1]
where Bi is a constant that represents the potential added by the ionic atmosphere. No justification for Bi being a constant is given. However, one can see that this is the case by considering that any spherical static charge distribution is subject to the mathematics of the shell theorem. The shell theorem says that no force is exerted on charged particles inside a sphere (of arbitrary charge) (http://hyperphysics.phy-astr.gsu.edu/hbase/electric/potsph.html). Since the ion atmosphere is assumed to be (time-averaged) spherically symmetric, with charge varying as a function of radius r, it may be represented as an infinite series of concentric charge shells. Therefore, inside the radius ai, the ion atmosphere exerts no force. If the force is zero, then the potential is a constant (by definition). In a combination of the continuously distributed model which gave the Poisson-Boltzmann equation and the model of the point charge, it is assumed that at the radius ai, there is a continuity of
By the definition of electric potential energy, the potential energy associated with the singled out ion in the ion atmosphere is[1] Notice that this only requires knowledge of the charge of the singled out ion and the potential of all the other ions. To calculate the potential energy of the entire electrolyte solution, one must use the multiple charge generalization for electric potential energy.[1] The Additional Electric Term to the Thermodynamic PotentialNondimensionalizationThis section was created without reference to the original paper and there are some errors in it (for instance, the ionic strength is off by a factor of two). Once these are rectified, this section should probably be moved to the nondimensionalization article and then be linked from here, since the nondimensional version of the Poisson-Boltzmann equation isn't necessary to understand the D&H theory. The differential equation is ready for solution (as stated above, the equation only holds for low concentrations): Using the Buckingham π theorem on this problem results in the following dimensionless groups:
Φ is called the reduced scalar electric potential field. R is called the reduced radius. The existing groups may be recombined to form two other dimensionless groups for substitution into the differential equation. The first is what could be called the square of the reduced inverse screening length, (κa)2. The second could be called the reduced central ion charge, Z0 (with a capital Z). Note that, though z0 is already dimensionless, without the substitution given below, the differential equation would still be dimensional. To obtain the nondimensionalized differential equation and initial conditions, use the π groups to eliminate For table salt in 0.01 M solution at 25°C, typical a typical value of (κa)2 is 0.0005636, while a typical value of Z0 is 7.017, highlighting the fact that, in low concentrations, (κa)2 is a target for a zero order of magnitude approximation such as perturbation analysis. Unfortunately, because of the boundary condition at infinity, regular perturbation does not work. The same boundary condition prevents us from finding the exact solution to the equations. Singular perturbation may work, however. Extensions of the theoryWarning: The notation in this section is (presently) different than in the rest of the article. A number of approaches have been proposed to extend the validity of the law to concentration ranges as commonly encountered in chemistry One such Extended Debye-Hückel Equation is given by: where The Extended Debye-Hückel Equation provides accurate results for μ ≤ 0.1 M. For solutions of greater ionic strengths, the Pitzer equation should be used. In these solutions the activity coefficient may actually increase with ionic strength. The Debye-Hückel Equation cannot be used in the solutions of surfactants where the presence of micelles influences on the electrochemical properties of the system (even rough judgement overestimates γ for ~50%). See also
References
Categories: Analytical chemistry | Electrochemistry |
|
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Debye-Hückel_equation". A list of authors is available in Wikipedia. |
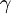 , and takes into account the interaction energy of ions in the solution.
, and takes into account the interaction energy of ions in the solution.
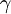 . The activity of some ion species C,
. The activity of some ion species C, 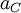 , is equal to a dimensionless measure of the concentration of C,
, is equal to a dimensionless measure of the concentration of C, ![\ [C] \,](images/math/2/d/8/2d8a6bfeb2b8fac8db6a995542ce79b1.png) multiplied by the activity coefficient of C,
multiplied by the activity coefficient of C, ![\ a_C = \gamma \frac{[C]}{[C_{\Theta}]}\,](images/math/3/a/b/3abc5bb5d0e00683dc75418ea4112a33.png)
![\ [C_{\Theta}]\,](images/math/0/9/8/09857a5bec7b36b4e8f46b50e9aea367.png) represents the concentration of the chosen standard state, e.g. 1 mol/kg if we work in molality.
represents the concentration of the chosen standard state, e.g. 1 mol/kg if we work in molality.
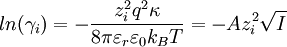
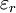 is the
is the 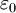 is the permittivity of free space
is the permittivity of free space



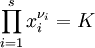
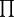 is a notation for multiplication
is a notation for multiplication
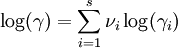 .
.
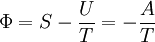
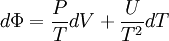
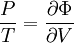 and
and
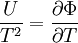 ,
,
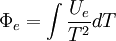
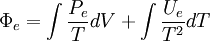 .
.
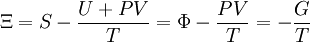 .
.
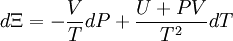 .
.
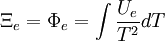 .
.
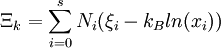 .
.
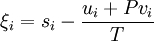 .
.
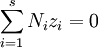 .
.
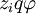 , where
, where 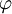 is the value of the scalar electric potential field at
is the value of the scalar electric potential field at 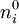 , by the corresponding
, by the corresponding 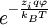 , where
, where 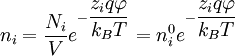
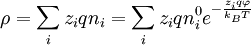 .
.
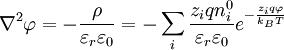 .
.
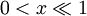 ) to create a linear differential equation (Hamann, Hamnett, and Vielstich. Electrochemistry. Wiley-VCH. section 2.4.2). D&H say that this approximation holds at large distances between ions,
) to create a linear differential equation (Hamann, Hamnett, and Vielstich. Electrochemistry. Wiley-VCH. section 2.4.2). D&H say that this approximation holds at large distances between ions,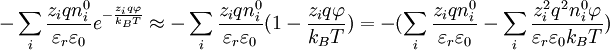 .
.
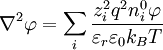 ,
,
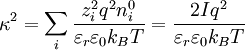 ,
,
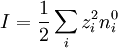 .
.
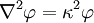 .
.
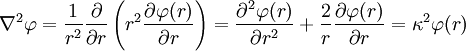 .
.
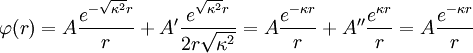 .
.
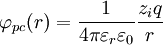 .
.
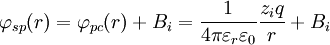 ,
,
 and its first derivative. Thus,
and its first derivative. Thus, ,
,
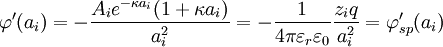 ,
,
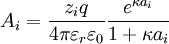 , and
, and
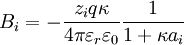 .
.
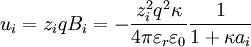
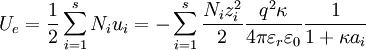
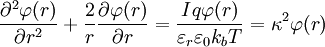
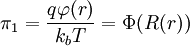
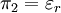
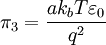
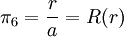
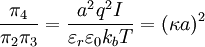
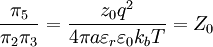
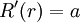 , then eliminate
, then eliminate 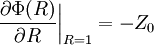
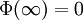
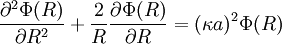
 is the integer charge of the ion (1 for H+, 2 for Mg2+ etc.),
is the integer charge of the ion (1 for H+, 2 for Mg2+ etc.),  is the ionic strength of the aqueous solution, and
is the ionic strength of the aqueous solution, and  is the size or effective diameter of the ion in angstrom. The effective hydrated radius of the ion, a is the radius of the ion and its closely bound water molecules. Large ions and less highly charged ions bind water less tightly and have smaller hydrated radii than smaller, more highly charged ions. Typical values are 3Å for ions such as H+,Cl-,CN-, and HCOO-. The effective diameter for the hydronium ion is 9Å.
is the size or effective diameter of the ion in angstrom. The effective hydrated radius of the ion, a is the radius of the ion and its closely bound water molecules. Large ions and less highly charged ions bind water less tightly and have smaller hydrated radii than smaller, more highly charged ions. Typical values are 3Å for ions such as H+,Cl-,CN-, and HCOO-. The effective diameter for the hydronium ion is 9Å. 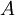 and
and 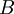 are constants with values of respectively 0.5085 and 0.3281 at 25°C in water
are constants with values of respectively 0.5085 and 0.3281 at 25°C in water 

