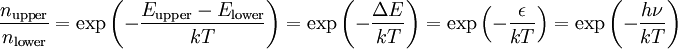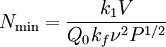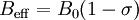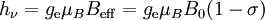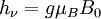To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Electron paramagnetic resonanceElectron paramagnetic resonance (EPR) or electron spin resonance (ESR) spectroscopy is a technique for studying chemical species that have one or more unpaired electrons, such as organic and inorganic free radicals or inorganic complexes possessing a transition metal ion. The basic physical concepts of EPR are analogous to those of nuclear magnetic resonance (NMR), but it is electron spins that are excited instead of spins of atomic nuclei. Because most stable molecules have all their electrons paired, the EPR technique is less widely used than NMR. However, this limitation to paramagnetic species also means that the EPR technique is one of great specificity, since ordinary chemical solvents and matrices do not give rise to EPR spectra. EPR was first observed in Kazan State University by the Soviet physicist Yevgeny Zavoisky in 1944, and was developed independently at the same time by Brebis Bleaney at Oxford University.
Product highlight
EPR theoryUnits and constants
Origin of an EPR signalEvery electron has a magnetic moment and spin quantum number s = 1/2, with magnetic components ms = +1/2 and ms = –1/2. In the presence of an external magnetic field with strength B0, the electron's magnetic moment aligns itself either parallel (ms = –1/2) or antiparallel (ms = +1/2) to the field, each alignment having a specific energy (see the Zeeman effect). The parallel alignment corresponds to the lower energy state, and the separation between it and the upper state is ΔE = geμBB0, where ge is the electron's so-called g-factor (see also the Landé g-factor). This equation implies that the splitting of the energy levels is directly proportional to the magnetic field's strength, as shown in the diagram below. An unpaired electron can move between the two energy levels by either absorbing or emitting electromagnetic radiation of energy ε = hν such that the resonance condition, ε = ΔE, is obeyed. Substituting in ε = hν and ΔE = geμBB0 leads to the fundamental equation of EPR spectroscopy: hν = geμBB0. Experimentally, this equation permits a large combination of frequency and magnetic field values, but the great majority of EPR measurements are made with microwaves in the 9000 – 10000 MHz (9 – 10 GHz) region, with fields corresponding to about 3500 G (0.35 T). See below for other field-frequency combinations. In principle, EPR spectra can be generated by either varying the photon frequency incident on a sample while holding the magnetic field constant, or doing the reverse. In practice, it is usually the frequency which is kept fixed. A collection of paramagnetic centers, such as free radicals, is exposed to microwaves at a fixed frequency. By increasing an external magnetic field, the gap between the ms = +1/2 and ms = −1/2 energy states is widened until it matches the energy of the microwaves, as represented by the double-arrow in the diagram above. At this point the unpaired electrons can move between their two spin states. Since there typically are more electrons in the lower state, due to the Maxwell-Boltzmann distribution (see below), there is a net absorption of energy, and it is this absorption which is monitored and converted into a spectrum. As an example of how hν = geμBB0 can be used, consider the case of a free electron, which has ge = 2.0023[1], and the simulated spectrum shown at the right in two different forms. For the microwave frequency of 9388.2 MHz, the predicted resonance position is a magnetic field of about B0 = hν / geμB = 0.3350 tesla = 3350 gauss, as shown. Note that while two forms of the same spectrum are presented in the figure, most EPR spectra are recorded and published only as first derivatives. Because of electron-nuclear mass differences, the magnetic moment of an electron is substantially larger than the corresponding quantity for any nucleus, so that a much higher electromagnetic frequency is needed to bring about a spin resonance with an electron than with a nucleus, at identical magnetic field strengths. For example, for the field of 3350 G shown at the right, spin resonance occurs near 9388.2 MHz for an electron compared to only about 14.3 MHz for 1H nuclei. (For NMR spectroscopy, the corresponding resonance equation is hν = gNμNB0 where gN and μN depend on the nucleus under study.) Maxwell-Boltzmann distributionIn practice, EPR samples consist of collections of many paramagnetic species, and not single isolated paramagnetic centers. If the population of radicals is in thermodynamic equilibrium, its statistical distribution is described by the Maxwell-Boltzmann equation where n upper is the number of paramagnetic centers occupying the upper energy state, k is the Boltzmann constant, and T is the temperature in kelvins. At 298 K, X-band microwave frequencies (ν ≈ 9.75 GHz) give nupper / nlower ≈ 0.998, meaning that the upper energy level has a smaller population than the lower one. Therefore, transitions from the lower to the higher level are more probable than the reverse, which is why there is a net absorption of energy. The sensitivity of the EPR method (i.e., the minimum number of detectable spins Nmin) depends on the photon frequency ν according to where k1 is a constant, V is the sample's volume, Q0 is the unloaded quality factor of the microwave cavity (sample chamber), kf is the cavity filling coefficient, and P is the microwave power in the spectrometer cavity. With kf and P being constants, Nmin ~ (Q0ν2) − 1, i.e., Nmin ~ ν − α, where α ≈ 1.5. In practice, α can change varying from 0.5 to 4.5 depending on spectrometer characteristics, resonance conditions, and sample size. In other words, the higher the spectrometer frequency the lower the detection limit (Nmin), meaning greater sensitivity. EPR spectral parametersIn real systems, electrons are normally not solitary, but are associated with one or more atoms. There are several important consequences of this:
The g factorKnowledge of the g-factor can give information about a paramagnetic center's electronic structure. An unpaired electron responds not only to a spectrometer's applied magnetic field B0, but also to any local magnetic fields of atoms or molecules. The effective field Beff experienced by an electron is thus written where σ includes the effects of local fields (σ can be positive or negative). Therefore, the hν = geμBBeff resonance condition (above) is rewritten as follows: The quantity ge(1 - σ) is denoted g and called simply the g-factor, so that the final resonance equation becomes This last equation is used to determine g in an EPR experiment by measuring the field and the frequency at which resonance occurs. If g does not equal ge the implication is that the ratio of the unpaired electron's spin magnetic moment to its angular momentum differs from the free electron value. Since an electron's spin magnetic moment is constant (approximately the Bohr magneton), then the electron must have gained or lost angular momentum through spin-orbit coupling. Because the mechanisms of spin-orbit coupling are well understood, the magnitude of the change gives information about the nature of the atomic or molecular orbital containing the unpaired electron. Hyperfine couplingSince the source of an EPR spectrum is a change in an electron's spin state, it might be thought that all EPR spectra would consist of a single line. However, the interaction of an unpaired electron, by way of its magnetic moment, with nearby nuclear spins, results in additional allowed energy states and, in turn, multi-lined spectra. In such cases, the spacing between the EPR spectral lines indicates the degree of interaction between the unpaired electron and the perturbing nuclei. The hyperfine coupling constant of a nucleus is directly related to the spectral line spacing and, in the simplest cases, is essentially the spacing itself. Two common mechanisms by which electrons and nuclei interact are Fermi-contact interaction and dipolar interaction. The former applies largely to the case of isotropic interactions (independent of sample orientation in a magnetic field) and the latter to the case of anisotropic interactions (spectra dependent on sample orientation in a magnetic field). Spin polarization is a third mechanism for interactions between an unpaired electron and a nuclear spin, being especially important for π-electron organic radicals, such as the benzene radical anion. The symbols "a" or "A" are used for isotropic hyperfine coupling constants while "B" is usually employed for anisotropic hyperfine coupling constants.[2] In many cases, the isotropic hyperfine splitting pattern for a radical freely tumbling in a solution (isotropic system) can be predicted.
While it is easy to predict the number of lines a radical's EPR spectrum should show, the reverse problem, unraveling a complex multi-line EPR spectrum and assigning the various spacings to specific nuclei, is more difficult. In the oft-encountered case of I = 1/2 nuclei (e.g., 1H, 19F, 31P), the line intensities produced by a population of radicals, each possessing M equivalent nuclei, will follow Pascal's triangle. For example, the spectrum at the right shows that the three 1H nuclei of the CH3 radical give rise to 2MI + 1 = 2(3)(1/2) + 1 = 4 lines with a 1:3:3:1 ratio. The line spacing gives a hyperfine coupling constant of aH = 23 G for each of the three 1H nuclei. Note again that the lines in this spectrum are first derivatives of absorptions. As a second example, consider the methoxymethyl radical, H2C(OCH3). The two equivalent methyl hydrogens will give an overall 1:2:1 EPR pattern, each component which is further split by the three methoxy hydrogens into a 1:3:3:1 pattern to give a total of 3 x 4 = 12 lines, a triplet of quartets. A simulation of the observed EPR spectrum is shown at the right, and agrees with the 12-line prediction and the expected line intensities. Note that the smaller coupling constant (smaller line spacing) is due to the three methoxy hydrogens, while the larger coupling constant (line spacing) is from the two hydrogens bonded directly to the carbon atom bearing the unpaired electron. It is often the case that coupling constants decrease in size with distance from a radical's unpaired electron, but there are some notable exceptions, such as the ethyl radical (CH2CH3). Resonance linewidth definitionResonance linewidths are defined in terms of the magnetic induction B, and its corresponding units, and are measured along the x axis of an EPR spectrum, from a line's center to a chosen reference point of the line. These defined widths are called halfwidths and possess some advantages: for asymmetric lines values of left and right halfwidth can be given. The halfwidth ΔBh is the distance measured from the line's center to the point in which absorption value has half of maximal absorption value in the center of resonance line. First inclination width ΔB1 / 2 is a distance from center of the line to the point of maximal absorption curve inclination. In practice, a full definition of linewidth is used. For symmetric lines, halfwidth ΔB1 / 2 = 2ΔBh, and full inclination width ΔBmax = 2ΔB1s EPR applicationsEPR spectroscopy is used in various branches of science, such as chemistry and physics, for the detection and identification of free radicals and paramagnetic centers. EPR is a sensitive, specific method for studying both radicals formed in chemical reactions and the reactions themselves. For example, when frozen water (solid H2O) is decomposed by exposure to high-energy radiation, radicals such as H, OH, and HO2 are produced. Such radicals can be identified and studied by EPR. Organic and inorganic radicals can be detected in electrochemical systems and in materials exposed to UV light. In many cases, the reactions to make the radicals and the subsequent reactions of the radicals are of interest, while in other cases EPR is used to provide information on a radical's geometry and the orbital of the unpaired electron. Medical and biological applications of EPR also exist. Although radicals are very reactive, and so do not normally occur in high concentrations in biology, special reagents have been developed to spin-label molecules of interest. These reagents are particularly useful in biological systems. Specially-designed nonreactive radical molecules can attach to specific sites in a biological cell, and EPR spectra can then give information on the environment of these so-called spin-label or spin-probes. EPR spectroscopy can only be applied to systems in which the balance between radical decay and radical formation keeps the free-radicals concentration above the detection limit of the spectrometer used. This can be a particularly severe problem in studying reactions in liquids. An alternative approach is to slow down reactions by studying samples held at cryogenic temperatures, such as 77 K (liquid nitrogen) or 4.2 K (liquid helium). An example of this work is the study of radical reactions in single crystals of amino acids exposed to x-rays, work that sometimes leads to activation energies and rate constants for radical reactions. EPR also has been used by archaeologists for the dating of teeth. Radiation damage over long periods of time creates free radicals in tooth enamel, which can then be examined by EPR and, after proper calibration, dated. Radiation-sterilized foods have been examined with EPR spectroscopy, the aim being to develop methods to determine if a particular food sample has been irradiated and to what dose. High-field-high-frequency measurementsHigh-field-high-frequency EPR measurements are sometimes needed to detect subtle spectroscopic details. However, for many years the use of electromagnets to produce the needed fields above 1.5 T was impossible, due principally to limitations of traditional magnet materials. The first multifunctional millimeter EPR spectrometer with a superconducting solenoid was described in the early 1970s by Prof. Y. S. Lebedev's group (Russian Institute of Chemical Physics, Moscow) in collaboration with L. G. Oranski's group (Ukrainen Physics and Technics Institute, Donetsk) which began working in the Institute of Problems of Chemical Physics, Chernogolovka around 1975.[1] Two decades later, a W-band EPR spectrometer was produced as a small commercial line by the German Bruker Company [2], initiating the expansion of W-band EPR techniques into medium-sized academic laboratories. Today there still are only a few scientific centers in the world capable of high-field-high-frequency EPR, among them are the Grenoble High Magnetic Field Laboratory in Grenoble, France, the National High Magnetic Field Laboratory in Tallahassee, USA, the IFW in Dresden, Germany, and the Institute of Physics of Complex Matter in Lausanne in Switzerland.
The EPR waveband is stipulated by the frequency or wavelength of a spectrometer's microwave source (see Table). EPR experiments often are conducted at X and, less commonly, Q bands, mainly due to the ready availability of the necessary microwave components (which originally were developed for radar applications). A second reason for widespread X and Q band measurements is that electromagnets can reliably generate fields up to about 1 tesla. However, the low spectral resolution over g-factor at these wavebands limits the study of paramagnetic centers with comparatively low anisotropic magnetic parameters. Measurements at ν > 40 GHz, in the millimeter wavelength region, offer the following advantages:
References
LiteratureMany good books and papers are available on the subject of EPR spectroscopy, including those listed here. Essentially all details in this article can be found in these.
See also
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Electron_paramagnetic_resonance". A list of authors is available in Wikipedia. |