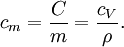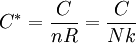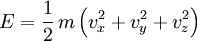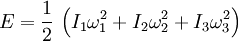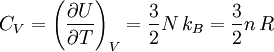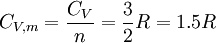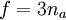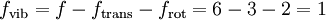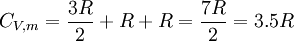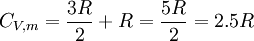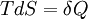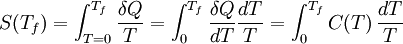To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Specific heat capacity
Specific heat capacity, also known simply as specific heat, is the measure of the heat energy required to increase the temperature of a unit quantity of a substance by a certain temperature interval. The term originated primarily through the work of Scottish physicist Joseph Black who conducted various heat measurements and used the phrase “capacity for heat.”[1] More heat energy is required to increase the temperature of a substance with high specific heat capacity than one with low specific heat capacity. For instance, eight times the heat energy is required to increase the temperature of an ingot of magnesium as is required for a lead ingot of the same mass. The specific heat of virtually any substance can be measured, including chemical elements, compounds, alloys, solutions, and composites. The symbols for specific heat capacity are either C or c depending on how the quantity of a substance is measured (see Symbols and standards below for usage rules). In the measurement of physical properties, the term “specific” means the measure is a bulk property (an intensive property), wherein the quantity of substance must be specified. For example, the heat energy required to raise water’s temperature one kelvin (equal to one degree Celsius) is approximately 4.2 joules per gram — the gram being the specified quantity. Scientifically, this measure would be expressed as c = 4.2 J g–1 K–1. Product highlight
Basic metrics of specific heat capacityUnit quantityWhen measuring specific heat capacity in science and engineering, the unit quantity of a substance is often in terms of mass: either the gram or kilogram, both of which are an SI unit. Especially in chemistry though, the unit quantity of specific heat capacity may also be the mole, which is a certain number of molecules or atoms. When the unit quantity is the mole, the term molar heat capacity may also be used to more explicitly describe the measure. Heat energyThe unit of measure for heat energy is usually the SI unit joule. The calorie however, is still often used in chemistry. Temperature intervalThe temperature interval in science, engineering, and chemistry is usually one kelvin or degree Celsius (both of which have the same magnitude). Other unitsIn the U.S., other units of measure for specific heat capacity are typically used in disciplines such as construction and civil engineering. There, the mass quantity is often the pound-mass, the unit of heat energy is the British thermal unit, and the temperature interval is the degree Fahrenheit. Basic equations
Factors that affect specific heat capacity
Symbols and standardsWhen mass is the unit quantity, the symbol for specific heat capacity is lowercase c. When the mole is the unit quantity, the symbol is uppercase C. Alternatively—especially in chemistry as opposed to engineering—the uppercase version for specific heat, C, may be used in combination with a suffix representing enthalpy (symbol: either H or h); specifically, when the mole is the unit quantity, the enthalpy suffix is uppercase H and when mass is the unit quantity, the suffix is lowercase h. The modern SI units for measuring specific heat capacity are either the joule per gram-kelvin (J g–1 K–1) or the joule per mole-kelvin (J mol–1 K–1). The various SI prefixes can create variations of these units (such as kJ kg–1 K–1 and kJ mol–1 K–1). Symbols for alternative units are as follows: pounds-mass (symbol: lb) for quantity, calories (symbol: cal) and British thermal units (symbol: BTU) for energy, and degree Fahrenheit (symbol: °F) for the increment of temperature. There are two distinctly different experimental conditions under which specific heat capacity is measured and these are denoted with a subscripted suffix modifying the symbols C or c. The specific heat of substances are typically measured under constant pressure (Symbols: Cp or cp). However, fluids (gases and liquids) are typically also measured at constant volume (Symbols: Cv or cv). Measurements under constant pressure produces greater values than those at constant volume because work must be performed in the former. This difference is particularly great in gases where values under constant pressure are typically 30% to 66.7% greater than those at constant volume. Thus, the symbols for specific heat capacity are as follows:
The ratio of the specific heats (or Heat capacity ratio) is usually denoted by γ (gamma). It is often used in equations, such as for calculating speed of sound in an ideal gas. The specific heat capacities of substances comprising molecules (distinct from the monatomic gases) are not fixed constants and vary somewhat depending on temperature. Accordingly, the temperature at which the measurement is made is usually also specified. Examples of two common ways to cite the specific heat of a substance are as follows: Water (liquid): cp = 4.1855 J g–1 K–1 (15 °C), and… The pressure at which specific heat capacity is measured is especially important for gases and liquids. The standard pressure was once virtually always “one standard atmosphere” which is defined as the sea level–equivalent value of precisely 101.325 kPa (760 Torr). In the case of water, 101.325 kPa is still typically used due to water’s unique role in temperature and physical standards. However, in 1982, the International Union of Pure and Applied Chemistry (IUPAC) recommended that for the purposes of specifying the physical properties of substances, “the standard pressure” should be defined as precisely 100 kPa (≈750.062 Torr).[4] Besides being a round number, this had a very practical effect: relatively few people live and work at precisely sea level; 100 kPa equates to the mean pressure at an altitude of about 112 meters (which is closer to the 194–meter, world–wide median altitude of human habitation). Accordingly, the pressure at which specific heat capacity is measured should be specified since one can not assume its value. An example of how pressure is specified is as follows: Water (gas): CvH = 28.03 J mol–1 K–1 (100 °C, 101.325 kPa) Note in the above specification that the experimental condition is at constant volume. Still, the pressure within this fixed volume is controlled and specified. Heat capacityHeat capacity (symbol: Cp) — as distinct from specific heat capacity — is the measure of the heat energy required to increase the temperature of an object by a certain temperature interval. Heat capacity is an extensive property because its value is proportional to the amount of material in the object; for example, a bathtub of water has a greater heat capacity than a cup of water. Heat capacity is usually expressed in units of J K–1 (or J/K), subject to the caveats and exceptions detailed in both Basic metrics of specific heat capacity and Symbols and standards, above. For instance, one could write that the gasoline in a 55-gallon drum has an average heat capacity of 347 kJ/K. The uncertainty of an object’s measured quantity is rarely better than one percent and this places an upper limit on the accuracy and precision of most stated values of heat capacity. Accordingly, it is usually unnecessary as a practical matter, to specify the defined state at which the measurement was made; e.g. “(25 °C, 100 kPa).” In most cases, it is assumed that the substance’s specific heat capacity is a published value and the object’s quantity is subject to such a sizable relative uncertainty that it renders this detail moot. An exception would be when an object has an accurately known or precisely defined quantity; e.g. “The heat capacity of the International Prototype Kilogram is 133 J/K (25 °C).” Another exception would be when the defined state varies significantly from standard conditions. Table of specific heat capacities
A Assuming an altitude of 194 meters above mean sea level (the world–wide median altitude of human habitation), an indoor temperature of 23 °C, a dewpoint of 9 °C (40.85% relative humidity), and 760 mm–Hg sea level–corrected barometric pressure (molar water vapor content = 1.16%). Specific heat capacity of building materials(Usually of interest to builders and solar designers)
Derivations of heat capacity and specific heat capacityDefinition of heat capacityHeat capacity is mathematically defined as the ratio of a small amount of heat δQ added to the body, to the corresponding small increase in its temperature dT: For thermodynamic systems with more than one physical dimension, the above definition does not give a single, unique quantity unless a particular infinitesimal path through the system’s phase space has been defined (this means that one needs to know at all times where all parts of the system are, how much mass they have, and how fast they are moving). This information is used to account for different ways that heat can be stored as kinetic energy (energy of motion) and potential energy (energy stored in force fields), as an object expands or contracts. For all real systems, the path through these changes must be explicitly defined, since the value of heat capacity depends on which path from one temperature to another, is chosen. Of particular usefulness in this context are the values of heat capacity for constant volume, CV, and constant pressure, CP. These will be defined below. Heat capacity of compressible bodiesThe state of a simple compressible body with fixed mass is described by two thermodynamic parameters such as temperature T and pressure p. Therefore as mentioned above, one may distinguish between heat capacity at constant volume, CV, and heat capacity at constant pressure, Cp: where
The increment of internal energy is the heat added and the work added: So the heat capacity at constant volume is The enthalpy is defined by H = U + PV. The increment of enthalpy is which, after replacing dU with the equation above and cancelling the PdV terms reduces to: So the heat capacity at constant pressure is Note that this last “definition” is a bit circular, since the concept of “enthalpy” itself was invented to be a measure of heat absorbed or produced at constant pressures (the conditions in which chemists usually work). As such, enthalpy merely accounts for the extra heat which is produced or absorbed by pressure-volume work at constant pressure. Thus, it is not surprising that constant-pressure heat capacities may be defined in terms of enthalpy, since “enthalpy” was defined in the first place to make this so. Specific heat capacityThe specific heat capacity of a material is which in the absence of phase transitions is equivalent to where
For gases, and also for other materials under high pressures, there is need to distinguish between different boundary conditions for the processes under consideration (since values differ significantly between different conditions). Typical processes for which a heat capacity may be defined include isobaric (constant pressure, dp = 0) or isochoric (constant volume, dV = 0) processes. The corresponding specific heat capacities are expressed as A related parameter to c is Dimensionless heat capacityThe dimensionless heat capacity of a material is where
Again, SI units shown for example. Theoretical modelsGas phaseThe specific heat of the gas is best conceptualized in terms of the degrees of freedom of an individual molecule. The different degrees of freedom correspond to the different ways in which the molecule may store energy. The molecule may store energy in its translational motion according to the familiar formula where m is the mass of the molecule and [vx,vy,vz] is velocity of the center of mass of the molecule. Each direction of motion constitutes a degree of freedom, so that there are three translational degrees of freedom. In addition, a molecule may have rotational motion. The kinetic energy of rotational motion is generally expressed as where I is the moment of inertia tensor of the molecule, and [ω1,ω2,ω3] is the angular velocity pseudovector (in a coordinate system aligned with the principle axes of the molecule). In general, then, there will be three additional degrees of freedom corresponding to the rotational motion of the molecule, (For linear molecules one of the inertia tensor terms vanishes and there are only two rotational degrees of freedom). The degrees of freedom corresponding to translations and rotations are called the “rigid” degrees of freedom, since they do not involve any deformation of the molecule. The motions of the atoms in a molecule which are not part of its gross translational motion or rotation may be classified as vibrational motions. It can be shown that if there are n atoms in the molecule, there will be as many as 3n − 3 − nr vibrational degrees of freedom, where nr is the number of rotational degrees of freedom. The actual number may be less due to various symmetries. If the molecule could be entirely described using classical mechanics, then we could use the theorem of equipartition of energy to predict that each degree of freedom would have an average energy in the amount of (1/2)kT where k is Boltzmann’s constant and T is the temperature. Our calculation of the heat content would be straightforward. Each molecule would be holding, on average, an energy of (f/2)kT where f is the total number of degrees of freedom in the molecule. The total internal energy of the gas would be (f/2)NkT where N is the total number of molecules. The heat capacity (at constant volume) would then be a constant (f/2)Nk , the specific heat capacity would be (f/2)k and the dimensionless heat capacity would be just f/2. The various degrees of freedom cannot generally be considered to obey classical mechanics. Classically, the energy residing in each degree of freedom is assumed to be continuous - it can take on any positive value, depending on the temperature. In reality, the amount of energy that may reside in a particular degree of freedom is quantized: It may only be increased and decreased in finite amounts. A good estimate of the size of this minimum amount is the energy of the first excited state of that degree of freedom above its ground state. For example, the first vibrational state of the HCl molecule has an energy of about 5.74 × 10–20 joule. If this amount of energy were deposited in a classical degree of freedom, it would correspond to a temperature of about 4156 K. If the temperature of the substance is so low that the equipartition energy of (1/2)kT is much smaller than this excitation energy, then there will be little or no energy in this degree of freedom. This degree of freedom is then said to be “frozen out". As mentioned above, the temperature corresponding to the first excited vibrational state of HCl is about 4156 K. For temperatures well below this value, the vibrational degrees of freedom of the HCL molecule will be frozen out. They will contain little energy and will not contribute to the heat content of the HCl gas. It can be seen that for each degree of freedom there is a critical temperature at which the degree of freedom “unfreezes” and begins to accept energy in a classical way. In the case of translational degrees of freedom, this temperature is that temperature at which the thermal wavelength of the molecules is roughly equal to the size of the container. For a container of macroscopic size (e.g. 10 cm) this temperature is extremely small and has no significance, since the gas will certainly liquify or freeze before this low temperature is reached. For any real gas we may consider translational degrees of freedom to always be classical and contain an average energy of (3/2)kT per molecule. The rotational degrees of freedom are the next to “unfreeze". In a diatomic gas, for example, the critical temperature for this transition is usually a few tens of kelvins. Finally, the vibrational degrees of freedom are generally the last to unfreeze. As an example, for diatomic gases, the critical temperature for the vibrational motion is usually a few thousands of kelvins. It should be noted that it has been assumed that atoms have no rotational or internal degrees of freedom. This is in fact untrue. For example, atomic electrons can exist in excited states and even the atomic nucleus can have excited states as well. Each of these internal degrees of freedom are assumed to be frozen out due to their relatively high excitation energy. Nevertheless, for sufficiently high temperatures, these degrees of freedom cannot be ignored. Monatomic gasIn the case of a monatomic gas such as helium under constant volume, if it assumed that no electronic or nuclear quantum excitations occur, each atom in the gas has only 3 degrees of freedom, all of a translational type. No energy dependence is associated with the degrees of freedom which define the position of the atoms. While, in fact, the degrees of freedom corresponding to the momenta of the atoms are quadratic, and thus contribute to the heat capacity. There are N atoms, each of which has 3 components of momentum, which leads to 3N total degrees of freedom. This gives: where
The following table shows experimental molar constant volume heat capacity measurements taken for each noble monatomic gas (at 1 atm and 25 °C):
It is apparent from the table that the experimental heat capacities of the monatomic noble gases agrees with this simple application of statistical mechanics to a very high degree. Diatomic gasIn the somewhat more complex case of an ideal gas of diatomic molecules, the presence of internal degrees of freedom are apparent. In addition to the three translational degrees of freedom, there are rotational and vibrational degrees of freedom. In general, the number of degrees of freedom, f, in a molecule with na atoms is 3na: Mathematically, there are a total of three rotational degrees of freedom, one corresponding to rotation about each of the axes of three dimensional space. However, in practice we shall only consider the existence of two degrees of rotational freedom for linear molecules. This approximation is valid because the moment of inertia about the internuclear axis is vanishingly small with respect other moments of inertia in the molecule (this is due to the extremely small radii of the atomic nuclei, compared to the distance between them in a molecule). Quantum mechanically, it can be shown that the interval between successive rotational energy eigenstates is inversely proportional to the moment of inertia about that axis. Because the moment of inertia about the internuclear axis is vanishingly small relative to the other two rotational axes, the energy spacing can be considered so high that no excitations of the rotational state can possibly occur unless the temperature is extremely high. We can easily calculate the expected number of vibrational degrees of freedom (or vibrational modes). There are three degrees of translational freedom, and two degrees of rotational freedom, therefore Each rotational and translational degree of freedom will contribute R/2 in the total molar heat capacity of the gas. Each vibrational mode will contribute R to the total molar heat capacity, however. This is because for each vibrational mode, there is a potential and kinetic energy component. Both the potential and kinetic components will contribute R/2 to the total molar heat capacity of the gas. Therefore, we expect that a diatomic molecule would have a molar constant-volume heat capacity of where the terms originate from the translational, rotational, and vibrational degrees of freedom, respectively. The following is a table of some molar constant-volume heat capacities of various diatomic gasses
From the above table, clearly there is a problem with the above theory. All of the diatomics examined have heat capacities that are lower than those predicted by the Equipartition Theorem, except Br2. However, as the atoms composing the molecules become heavier, the heat capacities move closer to their expected values. One of the reasons for this phenomenon is the quantization of vibrational, and to a lesser extent, rotational states. In fact, if it is assumed that the molecules remain in their lowest energy vibrational state because the inter-level energy spacings are large, the predicted molar constant volume heat capacity for a diatomic molecule becomes which is a fairly close approximation of the heat capacities of the lighter molecules in the above table. If the quantum harmonic oscillator approximation is made, it turns out that the quantum vibrational energy level spacings are actually inversely proportional to the square root of the reduced mass of the atoms composing the diatomic molecule. Therefore, in the case of the heavier diatomic molecules, the quantum vibrational energy level spacings become finer, which allows more excitations into higher vibrational levels at a fixed temperature. Solid phase
For matter in a crystalline solid phase, the Dulong-Petit law, which was discovered empirically, states that the dimensionless specific heat capacity assumes the value 3R. Indeed, for solid metallic chemical elements at room temperature, heat capacities range from about 2.8 to 3.4 (beryllium being a notable exception at 2.0). The theoretical maximum heat capacity for larger and larger multi-atomic gases at higher temperatures, also approaches the Dulong-Petit limit of 3R, so long as this is calculated per mole of atoms, not molecules. The reason is that gases with very large molecules, in theory have almost the same high-temperature heat capacity as solids, lacking only the (small) heat capacity contribution that comes from potential energy that cannot be stored between separate molecules in a gas. The Dulong-Petit “limit” results from the equipartition theorem, and as such is only valid in the classical limit of a microstate continuum, which is a high temperature limit. For light and non-metallic elements, as well as most of the common molecular solids based on carbon compounds at standard ambient temperature, quantum effects may also play an important role, as they do in multi-atomic gases. These effects usually combine to give heat capacities lower than 3 R per mole of atoms in the solid, although heat capacities calculated per mole of molecules in molecular solids may be more than 3 R. For example, the heat capacity of water ice at the melting point is about 4.6 R per mole of molecules, but only 1.5 R per mole of atoms. The lower number results from the “freezing out” of possible vibration modes for light atoms at suitably low temperatures, just as in many gases. These effects are seen in solids more often than liquids: for example the heat capacity of liquid water is again close to the theoretical 3 R per mole of atoms of the Dulong-Petit theoretical maximum. For a more modern and precise analysis of the heat capacities of solids, especially at low temperatures, it is useful to use the idea of phonons. See Debye model. Heat capacity at absolute zeroFrom the definition of entropy we can calculate the absolute entropy by integrating from zero temperature to the final temperature Tf The heat capacity must be zero at zero temperature in order for the above integral not to yield an infinite absolute entropy, thus violating the third law of thermodynamics. One of the strengths of the Debye model is that (unlike the preceding Einstein model) it predicts an approach of heat capacity toward zero as zero temperature is approached, and also predicts the proper mathematical form of this approach. See alsoReferences
Categories: Thermodynamic properties | Heat |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Specific_heat_capacity". A list of authors is available in Wikipedia. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
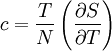
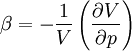
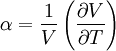


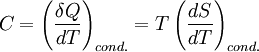
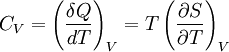
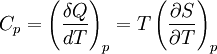
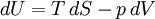
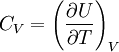
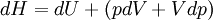
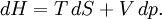
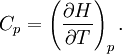
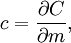
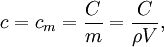
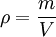 is the density of the material.
is the density of the material.
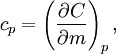
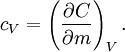
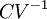 , the
, the 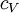 for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript
for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript 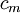 . Of course, from the above relationships, for solids one writes
. Of course, from the above relationships, for solids one writes