Designing new materials from 'small' data
Advertisement


Finding new functional materials is always tricky. But searching for very specific properties among a relatively small family of known materials is even more difficult.
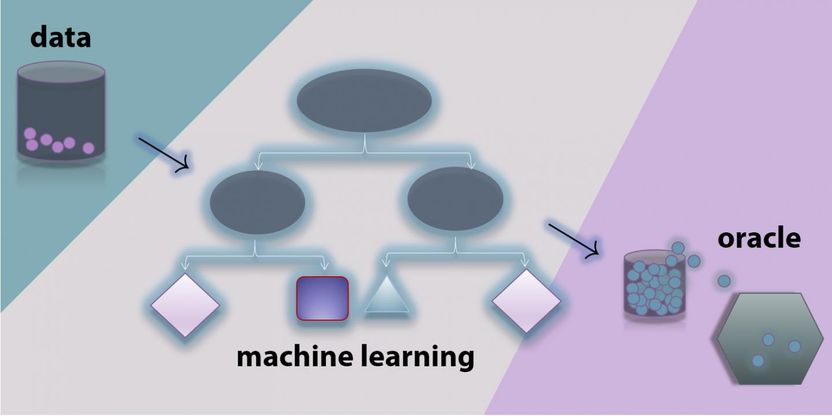
This is the novel data science approach using machine learning to find promising materials from small data.
James Rondinelli
But a team from Northwestern Engineering and Los Alamos National Laboratory found a workaround. The group developed a novel workflow combining machine learning and density functional theory calculations to create design guidelines for new materials that exhibit useful electronic properties, such as ferroelectricity and piezoelectricity.
Few layered materials have these qualities in certain geometries -- crucial for developing solutions to electronics, communication, and energy problems -- meaning there was very little data from which to formulate the guidelines using traditional research approaches.

"When others look for new materials, typically they look in places where they have a lot of data from similar materials. It's not necessarily easy by any means, but we do know how to distill information from large datasets," said James M. Rondinelli, assistant professor of materials science and engineering in the McCormick School of Engineering. "When you don't have a lot of information, learning from the data becomes a difficult problem."
Supported by funding from the National Science Foundation and the Laboratory Directed Research and Development Program through Los Alamos, Rondinelli's group focused on a class of two-dimensional complex oxides -- or Ruddlesden-Popper oxides. These materials exhibit many technology-enabling properties, such as ferroelectricity and piezoelectricity, and can be interfaced with traditional semiconductor materials found in today's electronic devices.
"In this family, the data set is puny. Currently, there are only around 10 to 15 materials that are known with the desired properties," Rondinelli said. "This is not much data to work with. Traditionally data science is used for big data problems where there is less of a need for domain knowledge."
"Despite the small data nature of the problem," Balachandran added, "our approach worked because we were able to combine our understanding of these materials (domain knowledge) with the data to inform the machine learning."
Therefore, the group began building a database of known materials and using machine learning, a subfield of computer science that builds algorithms capable of learning from data and then using that learning to make better predictions. "With machine learning, we are able to identify chemical compositions that are likely candidates for the material you want to develop," he said.
Of the more than 3,000 possible materials investigated, the data science approach found more than 200 with promising candidates. Next, the team applied several types of rigorous quantum mechanical calculations. This assessed the atomic structures of the potential materials and checked their stability.
"We wondered: Would the material have the predicted structure? Does it have electric polarization? Can it be made in a laboratory?" Rondinelli added.
This work narrowed the possibilities to 19, which were recommended for immediate experimental synthesis. Yet there are likely many more possibilities among the 200 candidates.
Typically, when developing new materials, the number of possibilities is too large to explore and develop each one. The process of screening potential materials is very expensive, and scientists must be selective in their investments.
"Our work has the potential to help save enormous amounts of time and resources," Balachandran said. "Instead of exploring all possible materials, only those materials that have the potential to be promising will be recommended for experimental investigation."
Original publication

Other news from the department science





















Most read news












More news from our other portals





























See the theme worlds for related content
Topic world Synthesis
Chemical synthesis is at the heart of modern chemistry and enables the targeted production of molecules with specific properties. By combining starting materials in defined reaction conditions, chemists can create a wide range of compounds, from simple molecules to complex active ingredients.

Topic world Synthesis
Chemical synthesis is at the heart of modern chemistry and enables the targeted production of molecules with specific properties. By combining starting materials in defined reaction conditions, chemists can create a wide range of compounds, from simple molecules to complex active ingredients.