The fast dance of electron spins
Chemists investigate the interactions of metal complexes and light
Advertisement



Metal complexes show a fascinating behavior in their interactions with light, which for example is utilized in organic light emitting diodes, solar cells, quantum computers, or even in cancer therapy. In many of these applications, the electron spin, a kind of inherent rotation of the electrons, plays an important role. Recently, the chemists Sebastian Mai and Leticia González from the Faculty of Chemistry of the University of Vienna succeeded in simulating the extremely fast spin flip processes that are triggered by the light absorption of metal complexes.
When a molecule is hit by light, in many cases a so-called "photoinduced" reaction is initiated. This can be thought of as the interplay of electron motion and nuclear motion. First, the absorption of light energetically "excites" the electrons, which for instance can weaken some of the bonds. Subsequently, the much heavier nuclei start moving. If at a later point in time the nuclei assume a favorable constellation with respect to each other, the electrons can switch from one orbit to another one. Controlled by the physical effect of "spin-orbit coupling" the electron spin can flip in the same moment.
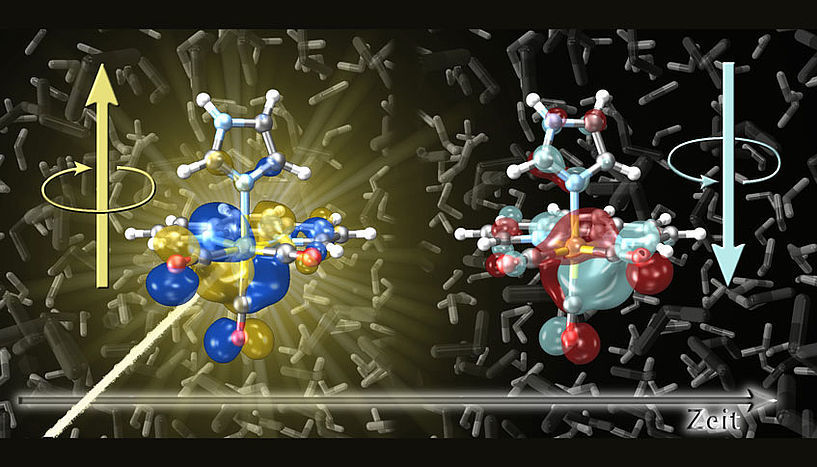
The extremely fast spin flip processes that are triggered by the light absorption of metal complexes were stimulated in the investigation.
© Sebastian Mai
This interplay of motion is the reason why spin-flip processes in molecules typically take quite long. However, computer simulations have shown that this is not the case in some metal complexes. For example, in the examined rhenium complex the spin-flip process already takes place within ten femtoseconds, even though in this short time the nuclei are virtually stationary—even light moves only three thousandths of a millimeter within this time. This knowledge is particularly useful for the precise control of electron spins, as, e.g., in quantum computers.
Investigation is based on enormous computer power
One of the biggest difficulties during the investigation was the huge amount of computer power that was required for the simulations. Although for small organic molecules one can nowadays carry out very accurate simulations already with a modest amount of computational effort, metal complexes present a much bigger challenge. Among other reasons, this is due to the large number of atoms, electrons, and solvent molecules that need to be included in the simulations, but also because the electron spin can only be accurately described with equations from relativity theory. Altogether, the scientists from the Institute of Theoretical Chemistry spent almost one million computer hours at the Austrian super computer "Vienna Scientific Cluster" in the course of their study. This is equivalent to about 100 years of computer time on a typical personal computer.
Original publication



Other news from the department science



























































