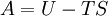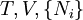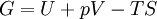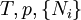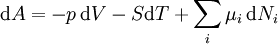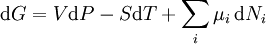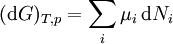To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Thermodynamic free energy
Additional recommended knowledgeIn thermodynamics, the term thermodynamic free energy is a measure of the amount of mechanical (or other) work that can be extracted from a system, and is helpful in engineering applications. It is a subtraction of the entropy of a system ("useless energy") from the total energy, yielding a thermodynamic state function which represents the "useful energy". OverviewIn short, free energy is that portion of any First-Law energy that is available for doing thermodynamic work; i.e., work mediated by thermal energy. Since free energy is subject to irreversible loss in the course of such work and First-Law energy is always conserved, it is evident that free energy is an expendable, Second-Law kind of energy that can make things happen within finite amounts of time. In solution chemistry and biochemistry, the Gibbs free energy change (denoted by ΔG) is commonly used merely as a surrogate for (−T times) the entropy produced by spontaneous chemical reactions in situations where there is no work done; or at least no "useful" work; i.e., other than pdV. As such, it serves as a particularization of the second law of thermodynamics, giving it the physical dimensions of energy, even though the inherent meaning in terms of entropy would be more to the point. The free energy functions are Legendre transforms of the internal energy. For processes involving a system at constant pressure p and temperature T, the Gibbs free energy is the most useful because, in addition to subsuming any entropy change due merely to heat flux, it does the same for the pdV work needed to "make space for additional molecules" produced by various processes. (Hence its utility to solution-phase chemists, including biochemists.) The Helmholtz free energy has a special theoretical importance since it is proportional to the logarithm of the partition function for the canonical ensemble in statistical mechanics. (Hence its utility to physicists; and to gas-phase chemists and engineers, who do not want to ignore pdV work.) The (historically earlier) Helmholtz free energy is defined as A = U − TS, where U is the internal energy, T is the absolute temperature, and S is the entropy. Its change is equal to the amount of reversible work done on, or obtainable from, a system at constant T. Thus its appellation "work content", and the designation A from arbeit, the German word for work. Since it makes no reference to any quantities involved in work (such as p and V), the Helmholtz function is completely general: its decrease is the maximum amount of work which can be done by a system, and it can increase at most by the amount of work done on a system. The Gibbs free energy G = H − TS, where H is the enthalpy. (H = U + pV, where p is the pressure and V is the volume.) There has been historical controversy:
Since both fields use both functions, a compromise has been suggested, using A to denote the Helmholtz function, with G for the Gibbs function. While A is preferred by IUPAC, F is sometimes still in use, and the correct free energy function is often implicit in manuscripts and presentations. ApplicationThe experimental usefulness of these functions is restricted to conditions where certain variables (T, and V or external p) are held constant, although they also have theoretical importance in deriving Maxwell relations. Work other than pdV may be added, e.g., for electrochemical cells, or f ˑdx work in elastic materials and in muscle contraction. Other forms of work which must sometimes be considered are stress-strain, magnetic, as in adiabatic demagnetization used in the approach to absolute zero, and work due to electric polarization. These are described by tensors. In most cases of interest there are internal degrees of freedom and processes, such as chemical reactions and phase transitions, which create entropy. Even for homogeneous "bulk" materials, the free energy functions depend on the (often suppressed) composition, as do all proper thermodynamic potentials (extensive functions), including the internal energy.
Ni is the number of molecules (alternatively, moles) of type i in the system. If these quantities do not appear, it is impossible to describe compositional changes. The differentials for reversible processes are (assuming only pV work) where μi is the chemical potential for the i-th component in the system. The second relation is especially useful at constant T and p, conditions which are easy to achieve experimentally, and which approximately characterize living creatures. Any decrease in the Gibbs function of a system is the upper limit for any isothermal, isobaric work that can be captured in the surroundings, or it may simply be dissipated, appearing as T times a corresponding increase in the entropy of the system and/or its surrounding. See alsoCategories: Free energy | Thermodynamics |
|||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Thermodynamic_free_energy". A list of authors is available in Wikipedia. | |||||||||||||||||||||