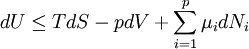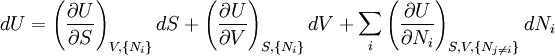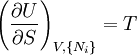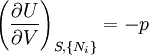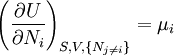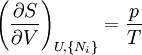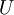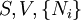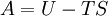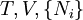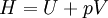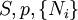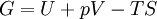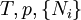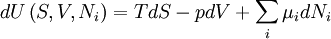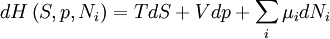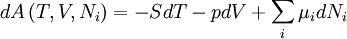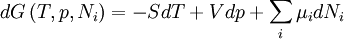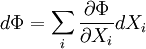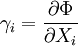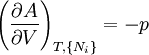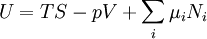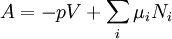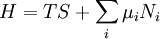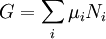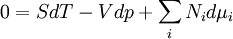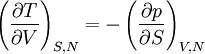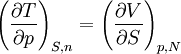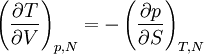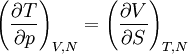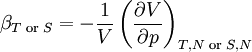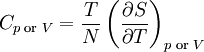To use all functions of this page, please activate cookies in your browser.
My watch list
my.chemeurope.com
my.chemeurope.com
With an accout for my.chemeurope.com you can always see everything at a glance – and you can configure your own website and individual newsletter.
- My watch list
- My saved searches
- My saved topics
- My newsletter
Thermodynamic equations
In thermodynamics, there are a large number of equations relating the various thermodynamic quantities. In chemical thermodynamics, which is a sub-branch of thermodynamics, for example, there are millions of useful equations. In particular, the structure of chemical thermodynamics is based on the first two laws of thermodynamics. Starting from the first and second laws of thermodynamics, four equations called the "fundamental equations of Gibbs" can be derived. From these four, more than fifty million equations, relating the thermodynamic properties of the thermodynamic system can be derived using relatively simple mathematics. This outlines the mathematical framework of chemical thermodynamics.[1] Some of the more commonly used thermodynamic equations are described below: Product highlight
IntroductionThe first thermodynamic equation, arguably, is the definition of "work" or weight lifted through a height, as defined in 1824 by French physicist Sadi Carnot. Technically, Carnot used the phrase motive power, which is what we currently refer to as work. In the footnotes to his famous On the Motive Power of Fire he states “We use here the expression motive power to express the useful effect that a motor is capable of producing. This effect can always be likened to the elevation of a weight to a certain height. It has, as we know, as a measure, the product of the weight multiplied by the height to which it is raised.” In this manner, Carnot is actually referring to "motive power" in the same manner we currently define "work". Hence, if we include a unit of time in Carnot's definition, we would then have the modern-day definition for power: During the latter half of the 19th century, physicists such as Rudolf Clausius, Peter Guthrie Tait, and Willard Gibbs worked to develop the concept of a thermodynamic system and the correlative energetic laws which govern its associated processes. The equilibrium state of a thermodynamic system is described by specifying its "state". The state of a thermodynamic system is specified by a number of extensive quantities, the most familiar of which are volume, internal energy, and the amount of each constituent particle (particle numbers). Extensive parameters are properties of the entire system, as contrasted with intensive parameters which can be defined at a single point, such as temperature and pressure. The extensive parameters (except entropy) are generally conserved in some way as long as the system is "insulated" to changes to that parameter from the outside. The truth of this statement for volume is trivial, for particles one might say that the total particle number of each atomic element is conserved. In the case of energy, the statement of the conservation of energy is known as the first law of thermodynamics. A thermodynamic system is in equilibrium when it is no longer changing in time. This may happen in a very short time, or it may happen with glacial slowness. A thermodynamic system may be composed of many subsystems which may or may not be "insulated" from each other with respect to the various extensive quantities. If we have a thermodynamic system in equilibrium in which we relax some of its constraints, it will move to a new equilibrium state. The thermodynamic parameters may now be thought of as variables and the state may be thought of as a particular point in a space of thermodynamic parameters. The change in the state of the system can be seen as a path in this state space. This change is called a thermodynamic process. Thermodynamic equations are now used to express the relationships between the state parameters at these different equilibrium state. The concept which governs the path that a thermodynamic system traces in state space as it goes from one equilibrium state to another is that of entropy. The entropy is first viewed as an extensive function of all of the extensive thermodynamic parameters. If we have a thermodynamic system in equilibrium, and we release some of the extensive constraints on the system, there are many equilibrium states that it could move to consistent with the conservation of energy, volume, etc. The second law of thermodynamics specifies that the equilibrium state that it moves to is in fact the one with the greatest entropy. Once we know the entropy as a function of the extensive variables of the system, we will be able to predict the final equilibrium state. (Callen, 1985) NotationSome of the most common thermodynamic quantites are:
The laws of thermodynamicsThe behavior of a thermodynamic system is summarized in what are known as the four laws of thermodynamics, which concisely are:
The zeroth law states that if two systems are equilibrium with a third, they are in equilibrium with each other. This law is actually the last of the laws to be formulated, and it essentially states that the equilibrium relationship is an equivalence relation. The first law is the law of conservation of energy is, in short form, The second law summarizes the tendency in which intensive thermodynamic properties, such as pressure, temperature, etc., have a tendency to equalize as time goes by, or: The third law of thermodynamics states that at the absolute zero of temperature, the entropy is at a minimum and all thermodynamic processes cease. The fourth law of thermodynamics is not yet an agreed upon law (many supposed variations exist); historically, however, the Onsanger reciprocal relations have been frequently referred to as the fourth law. The fundamental equation
The first and second law of thermodynamics are the most fundamental equations of thermodynamics. They may be combined into what is known as a "fundamental equation" which describes all of the thermodynamic properties of a system. As a simple example, consider a system composed of a number of p different types of particles. The fundamental equation may be expressed in terms of the internal energy as: Some important aspects of this equation should be noted: (Alberty, 2001), (Balian, 2003), (Callen, 1985)
Thermodynamic potentialsBy the principle of minimum energy, the second law can be restated by saying that for a fixed entropy, when the constraints on the system are relaxed, the internal energy assumes a minimum value. This will require that the system be connected to its surroundings, since otherwise the energy would remain constant. By the principle of minimum energy, there are a number of other state functions which may be defined which have the dimensions of energy and which are minimized according to the second law under certain conditions other than constant entropy. These are called thermodynamic potentials. The four most common thermodynamic potentials are:
After each potential is shown its "natural variables". These variables are important because if the thermodynamic potential is expressed in terms of its natural variables, then it will contain all of the thermodynamic relationships necessary to derive any other relationship. In other words, it too will be a fundamental equation. For the above four potentials, the fundamental equations are expressed as: First order equations
Just as with the internal energy version of the fundamental equation, the chain rule can be used on the above equations to find p+2 equations of state with respect to the particular potential. If Φ is a thermodynamic potential, then the fundamental equation may be expressed as: where the Xi are the natural variables of the potential. If γi is conjugate to Xi then we have the equations of state for that potential, one for each set of conjugate variables. Only one equation of state will not be sufficient to reconstitute the fundamental equation. All equations of state will be needed to fully characterize the thermodynamic system. Note that what is commonly called "the equation of state" is just the "mechanical" equation of state involving the Helmholtz potential and the volume: For an ideal gas, this becomes the familiar PV=NkT. Euler integrals
Because all of natural variables of the internal energy U are extensive quantities, it follows from Euler's homogeneous function theorem that Substituting into the expressions for the other main potentials we have the following expressions for the thermodynamic potentials: Note that the Euler integrals are sometimes also referred to as fundamental equations. Gibbs Duhem relationship
Differentiating the Euler equation for the internal energy and combining with the fundamental equation for internal energy, it follows that: which is known as the Gibbs-Duhem relationship. The Gibbs-Duhem is a relationship among the intensive parameters of the system. It follows that for a simple system with r components, there will be r+1 independent parameters, or degrees of freedom. For example, a simple system with a single component will have two degrees of freedom, and may be specified by only two parameters, such as pressure and volume for example. The law is named after Josiah Gibbs and Pierre Duhem. Second order equationsThere are many relationships that follow mathematically from the above basic equations. See Exact differential for a list of mathematical relationships. Many equations are expressed as second derivatives of the thermodynamic potentials (see Bridgman equations). Maxwell relationsMaxwell relations are equalities involving the second derivatives of thermodynamic potentials with respect to their natural variables. They follow directly from the fact that the order of differentiation does not matter when taking the second derivative. The four most common Maxwell relations are: Material propertiesSecond derivatives of thermodynamic potentials generally describe the response of the system to small changes. The number of second derivatives which are independent of each other is relatively small, which means that most material properties can be described in terms of just a few "standard" properties. For the case of a single component system, there are three properties generally considered "standard" from which all others may be derived:
These properties are seen to be the three possible second derivative of the Gibbs free energy with respect to temperature and pressure. Notes
References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| This article is licensed under the GNU Free Documentation License. It uses material from the Wikipedia article "Thermodynamic_equations". A list of authors is available in Wikipedia. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



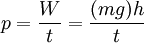
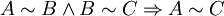
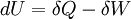
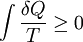
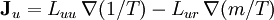 ;
;
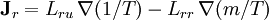 .
.
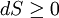 , where S is the entropy of the system. The entropy is generally related to the heat term by
, where S is the entropy of the system. The entropy is generally related to the heat term by